Lupus nephritis (LN) refers to kidney damage caused by systemic lupus erythematosus (SLE). More than 50% of SLE patients develop LN, making it one of the most critical and life-threatening organ involvements in SLE. It is also a significant cause of end-stage renal disease (ESRD).
Pathogenesis
The development of LN involves multiple factors, including genetic susceptibility, environmental triggers, and immune abnormalities. The formation and deposition of immune complexes in the kidneys is the primary mechanism underlying LN. These deposited immune complexes activate the complement system, leading to the infiltration of inflammatory cells, activation of coagulation factors, and release of inflammatory mediators, which ultimately result in kidney damage.
Pathology
The pathological manifestations of LN are diverse. In 2018, the International Society of Nephrology (ISN) and the Renal Pathology Society (RPS) revised the pathological classification of LN, as shown in Table 1.
Table 1 2018 ISN/RPS lupus nephritis (LN) pathological classification
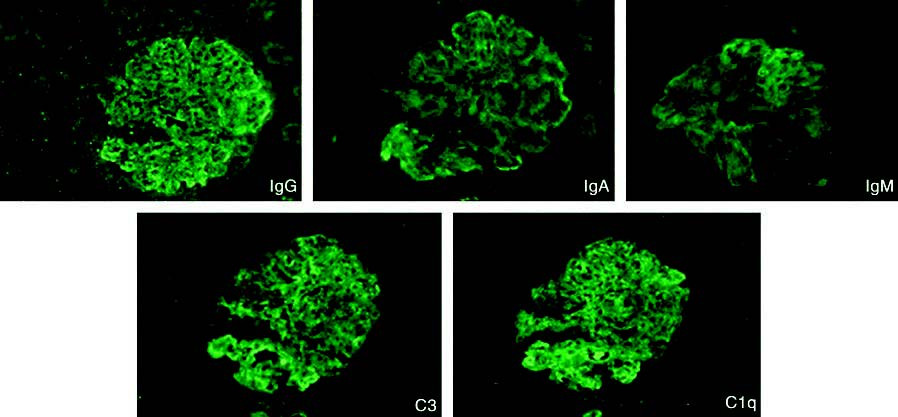
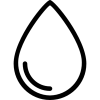
LN can affect the tubulointerstitial and vascular compartments in addition to the glomeruli. Specific subtypes include crescentic glomerulonephritis, thrombotic microangiopathy, and lupus podocytopathy. A hallmark immunopathological feature is the presence of glomerular deposits of IgA, IgG, IgM, C3, C4, and C1q, which is referred to as the "full house" pattern. Pathological types may change as the disease progresses or after treatment.
Figure 1 "Full house" pattern in lupus nephritis immunofluorescence
Clinical Manifestations
The extrarenal manifestations of LN are consistent with those of systemic lupus erythematosus. Renal manifestations of LN vary widely and can include asymptomatic hematuria and/or proteinuria, nephrotic syndrome, acute nephritic syndrome, or rapidly progressive nephritic syndrome.
Proteinuria is the most common manifestation and can range from mild to severe. Massive proteinuria and nephrotic syndrome are often observed in diffuse proliferative and/or membranous LN. Most patients exhibit microscopic hematuria, while gross hematuria is primarily seen in patients with loop necrosis or crescent formation. Hypertension is common and occurs more frequently in the presence of renal vascular lesions, sometimes even progressing to malignant hypertension.
Acute kidney injury may occur in diffuse proliferative LN, particularly in cases with severe endocapillary proliferative lesions and/or focal necrotizing crescentic glomerulonephritis. It may also be seen in patients with vasculitis or thrombotic microangiopathy. Patients with positive serum antiphospholipid antibodies are prone to thrombosis, which can further exacerbate kidney function deterioration.
Laboratory and Other Tests
Changes in urinary protein and red blood cells, complement levels, autoantibody titers (e.g., anti-dsDNA antibodies), serum albumin, and serum creatinine are closely related to LN activity and remission. Renal biopsy findings, including the activity and chronicity of lesions as well as the overall lupus activity, are valuable for diagnosing LN, guiding treatment, and predicting prognosis. In cases where patients show poor treatment response, relapse, or disease progression, repeat renal biopsy may be necessary for further evaluation.
Diagnosis and Differential Diagnosis
LN can be diagnosed in patients with SLE who exhibit renal involvement, such as persistent proteinuria (24-hour urinary protein >0.5 g/day, >++ on dipstick, or urine albumin-to-creatinine ratio >500 mg/g), with or without active urinary sediment (excluding urinary tract infection, defined as urine white blood cells >5/HPF or urine red blood cells >5/HPF) or casts (e.g., red blood cell casts or granular casts).
LN may be misdiagnosed as primary glomerular disease. Differentiation can be aided by identifying the presence of multi-system and multi-organ involvement, positive serum autoantibodies (e.g., ANA, anti-dsDNA, and anti-Sm antibodies), and renal biopsy findings. Differential diagnoses include IgA nephropathy, Henoch-Schönlein purpura nephritis, ANCA-associated vasculitis with renal involvement, and renal involvement in Sjögren's syndrome.
Treatment
The primary goals of lupus nephritis (LN) treatment are to control disease activity and prevent the progression of kidney damage. Treatment plans are individualized based on the overall activity of systemic lupus erythematosus (SLE), clinical manifestations, and pathological features. Long-term treatment typically involves an initial induction phase followed by maintenance therapy, along with regular monitoring of disease activity.
For proliferative LN (Class III or IV), induction therapy generally involves corticosteroids combined with immunosuppressive agents. Recommended regimens include corticosteroids combined with mycophenolate mofetil (MMF), cyclophosphamide, MMF plus belimumab, cyclophosphamide plus belimumab, or MMF plus calcineurin inhibitors. Once the disease stabilizes, patients transition to maintenance therapy. Induction therapy may include methylprednisolone pulse therapy at a dose of 0.25–0.5 g/day for 1–3 days, followed by oral prednisone at 0.5–1 mg/kg/day for 2–4 weeks, with gradual tapering to a maintenance dose of 5 mg/day. Corticosteroids are combined with immunosuppressive agents, such as:
- Intravenous cyclophosphamide (0.5–1.0 g/m2 monthly for 6 doses, or 0.5 g every 2 weeks for 6 doses)
- Oral cyclophosphamide (1–1.5 mg/kg/day)
- Mycophenolate mofetil (1.5–2.0 g/day, given in two divided doses)
If the response to induction therapy is suboptimal, further analysis is required to adjust the treatment regimen. Maintenance therapy often involves low-dose corticosteroids (prednisone 5 mg/day) combined with azathioprine (1.5–2 mg/kg/day) or mycophenolate mofetil (1–2 g/day, given in two divided doses). In cases of extensive cellular crescents or fibrinoid necrosis on renal biopsy, or severe extrarenal disease activity, intravenous methylprednisolone (15 mg/kg/day) may be used as pulse therapy once daily for 3 days per course.
For Class I, II, or V LN, patients with proteinuria below nephrotic levels may receive symptomatic and supportive treatment, such as ACE inhibitors/ARBs for proteinuria and blood pressure control, lipid-lowering therapy, anticoagulation, and hydroxychloroquine. The use of corticosteroids and immunosuppressive agents depends on extrarenal manifestations. If proteinuria increases or related complications worsen during treatment, corticosteroids combined with immunosuppressive agents may be required. For patients with nephrotic-range proteinuria, corticosteroids combined with immunosuppressive agents, such as prednisone with cyclophosphamide, mycophenolate mofetil, cyclosporine, or tacrolimus, are used. For Class I and II LN, electron microscopy is necessary to determine whether lupus podocytopathy is present.
During lupus treatment, monitoring for infections, osteoporosis, cardiovascular disease, malignancies, and pregnancy is essential. Multidisciplinary discussions may be required to develop comprehensive treatment plans when necessary.
Prognosis
LN can achieve long-term remission with treatment, but relapse is common after dose reduction or discontinuation of therapy. Some patients may experience gradual disease progression. The primary causes of death in LN patients include infections, cardiovascular and cerebrovascular diseases, and malignancies. In recent years, advancements in the diagnosis and management of LN have significantly improved outcomes, with 10-year survival rates now reaching 80–90%.