Lung cancer, also known as bronchogenic carcinoma, is defined by the World Health Organization (WHO) as a malignant tumor originating from the respiratory epithelial cells (bronchi, bronchioles, and alveoli). It is the most common primary malignant tumor of the lungs. Based on pathological types, lung cancer is classified into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). The peak incidence occurs between the ages of 55 and 65, with males being more affected than females, at a male-to-female ratio of approximately 2.1:1. Clinical symptoms are often insidious, with cough, expectoration, hemoptysis, and weight loss being the main manifestations. On chest X-rays, lung cancer typically appears as nodules, masses, or other lesions. Approximately 75% of patients are already in the advanced stages of lung cancer at the time of diagnosis, with an overall 5-year survival rate of around 20%. Therefore, improving survival rates requires a focus on early diagnosis and standardized treatment.
Epidemiology
Lung cancer is the leading cause of cancer-related deaths worldwide. According to WHO data (GLOBOCAN 2020), there were 2.2 million new lung cancer cases globally in 2020, accounting for 11.4% of all cancer cases (excluding non-melanoma skin cancers). Lung cancer was responsible for 1.8 million deaths, representing 18.0% of all cancer-related deaths. Over the past 20 years, the incidence and mortality rates of lung cancer among men in Western countries have declined, while they have continued to rise in developing countries. Mortality rates among women are still increasing in most parts of the world.
Etiology and Pathogenesis
The exact causes and mechanisms of lung cancer remain unclear, but evidence suggests associations with the following factors:
Smoking
Smoking is the most common cause of lung cancer, with approximately 85% of lung cancer patients having a history of smoking, including current and former smokers (defined as those who quit smoking at least 12 months before diagnosis). The risk of lung cancer increases significantly in individuals with a smoking history of 20-30 pack-years (defined as smoking one pack per day for 20-30 years). A recent study from the UK Biobank found that both prenatal tobacco exposure and postnatal smoking behaviors increase the risk of lung cancer, with earlier initiation of smoking associated with higher risk. Compared to non-smokers, smokers have an average 10-fold increased risk of developing lung cancer, and heavy smokers have a 10-25-fold increased risk. Smoking behavior acquired during childhood, adolescence, or adulthood increases the risk of lung cancer by 14-fold, 8-fold, and 5-fold, respectively, as well as the risk of lung cancer-related death. Former smokers have a lower risk of lung cancer than current smokers, but their risk remains 9 times higher than that of never-smokers. The risk decreases progressively with longer periods of smoking cessation. There is a clear relationship between smoking and lung cancer, with earlier initiation, longer duration, and higher intensity of smoking correlating with higher incidence and mortality rates of lung cancer.
Environmental tobacco smoke (ETS), also known as secondhand smoke or passive smoking, is another cause of lung cancer. The risk from ETS is lower than that of active smoking, but non-smokers who live with smokers for many years have a 20-30% increased risk of lung cancer, with the risk rising with the smoking intensity of their spouse. Tobacco is classified as a Group A carcinogen, and smoking is associated with an increased risk of all pathological types of lung cancer.
Occupational Carcinogens
Certain occupational environments contain numerous carcinogenic substances. Recognized carcinogens include asbestos, arsenic, bis(chloromethyl) ether, chromium, mustard gas, nickel, radon, cadmium, beryllium, polycyclic aromatic hydrocarbons, and radioactive substances such as radon and radon gas produced during the decay of uranium and radium. Other sources include ionizing radiation and microwave radiation. Silica and coal smoke are also confirmed lung carcinogens. These factors can increase the risk of lung cancer by 3-30 times. Since lung cancer development is a long process with a latency period of up to 20 years or more, many patients develop lung cancer long after ceasing exposure to carcinogens.
Air Pollution
Outdoor Pollution
Industrial emissions, vehicle exhaust, and other urban pollutants contain carcinogens such as benzo[a]pyrene, arsenic oxides, radioactive substances, nickel, chromium compounds, SO2, NO, and unburned aliphatic hydrocarbons. PM2.5, also known as fine particulate matter, is one of the most harmful pollutants in the atmosphere and is associated with lung cancer incidence and mortality. The risk increases with higher PM2.5 concentrations. Data suggest that lung cancer incidence rates are significantly higher in urban areas than in rural areas.
Indoor Pollution
Indoor passive smoking, fuel combustion, and cooking processes can release carcinogens. Indoor exposure to coal smoke or its incomplete combustion products is a risk factor for lung cancer, particularly for adenocarcinoma in women. Cooking oil fumes released during cooking are also a noteworthy carcinogenic factor.
Ionizing Radiation
Ionizing radiation may be occupational or non-occupational and can result from external sources or internal exposure through inhalation of radioactive dust and gases. The effects vary with the type of radiation. For instance, the atomic bomb in Hiroshima released neutrons and alpha radiation, while Nagasaki only experienced alpha radiation; the former resulted in a higher risk of lung cancer. A 1978 report from the U.S. indicated that 49.6% of ionizing radiation exposure in the general population came from natural sources, while 44.6% was from medical exposure, including 36.7% from X-ray diagnostics.
Diet and Physical Activity
Studies show that low intake of fruits and vegetables in adulthood is associated with a higher risk of lung cancer. Individuals with low serum beta-carotene levels also have a higher risk of lung cancer. Conversely, moderate to high-intensity physical activity can reduce lung cancer risk by 13-30%.
Genetics and Genetic Alterations
Genetic factors play a significant role in lung cancer. For example, individuals with a family history of early-onset lung cancer (before age 60) have a twofold increased risk of developing lung cancer. At the same level of tobacco exposure, women have a higher risk of lung cancer than men. Only about 11% of heavy smokers develop lung cancer, suggesting that genetic susceptibility may play a role. Lung cancer likely results from external factors triggering internal mechanisms, leading to malignant cellular transformation and irreversible genetic changes. These include activation of proto-oncogenes, inactivation of tumor suppressor genes, activation of autocrine feedback loops, and inhibition of apoptosis. Lung cancer is a multistage process involving a series of genetic alterations, with the accumulation of these changes disrupting cell growth and differentiation control, leading to uncontrolled growth and malignancy. Key oncogenes associated with lung cancer include the HER family, RAS family, MYC family, ALK fusion genes, Sox genes, and MDM2 genes. Tumor suppressor genes associated with lung cancer include TP53, RB1, CDKN2A, NME1, and PTEN. Other molecular mechanisms involved in lung cancer include activation of growth factor signaling pathways, tumor angiogenesis, apoptosis inhibition, and immune evasion.
Other Factors
The American Cancer Society lists tuberculosis as a risk factor for lung cancer, with affected individuals having a 10-fold higher risk than the general population, primarily for adenocarcinoma. Chronic lung diseases such as COPD, sarcoidosis, idiopathic pulmonary fibrosis, and scleroderma, as well as viral and fungal infections (e.g., Aspergillus), may also contribute to lung cancer development.
Classification
Classification by Anatomical Location
Central Lung Cancer
This type originates in the segmental and suprasegmental bronchi and is most commonly associated with squamous cell carcinoma and small cell lung cancer.
Peripheral Lung Cancer
This type occurs in the infrasegmental bronchi and is more commonly associated with adenocarcinoma.
Classification by Histopathology
According to the 2021 WHO histological classification of lung tumors, lung cancer is divided into two main categories: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC is the most common type, accounting for approximately 85% of all lung cancer cases. Additionally, atypical adenomatous hyperplasia (AAH) and adenocarcinoma in situ (AIS) are classified as glandular precursor lesions.
Non-Small Cell Lung Cancer (NSCLC)
Squamous Cell Carcinoma (SCC)
Squamous cell carcinoma (SCC) includes squamous cell carcinoma, not otherwise specified (keratinizing, non-keratinizing, and basaloid squamous carcinoma), and lymphoepithelial carcinoma.
Typical SCC originates from the squamous metaplasia of bronchial epithelium and often shows keratinization and/or intercellular bridges. Non-keratinizing SCC lacks these features and requires immunohistochemistry to confirm squamous differentiation. Basaloid squamous carcinoma is defined by at least 50% basaloid carcinoma cells.
Lymphoepithelial carcinoma is a poorly differentiated SCC with varying degrees of lymphocyte and plasma cell infiltration. Over 90% of Asian cases are associated with Epstein-Barr virus (EBV), whereas the association is less common in Western populations. EBV positivity is confirmed by in situ hybridization for EBER. Immunohistochemical staining shows CK5/6, p40, and p63 positivity in cancer cells.
Squamous cell carcinoma typically arises from the mucosa of segmental or subsegmental bronchi and tends to grow into the bronchial lumen. Early symptoms often include bronchial narrowing, leading to atelectasis or obstructive pneumonia. The tumor tissue is prone to degeneration and necrosis, forming cavities or cancerous lung abscesses. It is more common in older males. SCC generally grows slowly, metastasizes late, and is more amenable to surgical resection, resulting in a relatively high 5-year survival rate. However, it is less sensitive to chemotherapy and radiotherapy compared to small cell lung cancer.
Adenocarcinoma
Minimally invasive adenocarcinoma (MIA) predominantly consists of a lepidic growth pattern. The tumor diameter is ≤3 cm, with the invasive component measuring ≤5 mm in the stroma. There is no evidence of bronchial, vascular, or pleural invasion, tumor necrosis, or aerogenous spread.
Invasive non-mucinous adenocarcinoma demonstrates evidence of glandular differentiation either morphologically or immunohistochemically. Subtypes are recorded based on the predominant pattern (≥5%), and classification into a dominant subtype is no longer required. Common subtypes include lepidic, acinar, papillary, micropapillary, and solid patterns, often presenting as a mixture of multiple subtypes.
The grading system for early-stage invasive non-mucinous adenocarcinoma was proposed by the International Association for the Study of Lung Cancer (IASLC). Grading is based on the predominant histological type and the proportion of high-grade structures:
- Grade 1 (Well-differentiated): Lepidic-predominant adenocarcinoma without high-grade components or with <20% high-grade components.
- Grade 2 (Moderately differentiated): Acinar or papillary-predominant adenocarcinoma without high-grade components or with <20% high-grade components.
- Grade 3 (Poorly differentiated): Any histological type of adenocarcinoma with ≥20% high-grade components.
Invasive mucinous adenocarcinoma accounts for 3% of lung adenocarcinomas. Tumor cells are goblet-shaped or columnar, rich in intracellular mucin, with small nuclei pushed to one side. Surrounding alveoli are often filled with mucin. Growth patterns include lepidic, acinar, papillary, micropapillary, solid, and cribriform types. It may coexist with non-mucinous adenocarcinoma. Immunohistochemistry shows CK7 positivity, partial CK20 and CDX2 positivity, and TTF-1 and Napsin A negativity.
Colloid adenocarcinoma is rare, accounting for 0.14%-0.25% of lung cancers. It is characterized by abundant extracellular mucin, which usually constitutes >50% of the tumor. Immunohistochemistry shows positivity for CDX-20, CK20, and Villin.
Fetal adenocarcinoma of the lung (FLAC) is rare, accounting for 0.1%-0.5% of lung cancers. Pathologically, it resembles fetal lung tubules rich in glycogen and lacking cilia. FLAC is divided into low-grade and high-grade subtypes:
- High-grade FLAC: Common in older males (60-70 years old), often with a history of heavy smoking. Serum AFP levels may be elevated, and most cases are diagnosed at stage III-IV.
- Low-grade FLAC: More common in young females (30-40 years old), often diagnosed at stage I-II.
Pulmonary enteric adenocarcinoma is rare, with histological features resembling colorectal adenocarcinoma. The tumor often forms regular or irregular tubular, papillary, or cribriform structures. Poorly differentiated cases may present as solid nests. Some tumors express intestinal differentiation markers (CK7, CK20, CDX-2, Villin, MUC2, and HNF4a), while others lack marker expression despite intestinal morphology. Clinical evaluation is necessary to rule out gastrointestinal adenocarcinoma.
Adenocarcinoma is the most common type of lung cancer, particularly in women. It primarily originates from bronchial mucosal glands and often presents as peripheral lung cancer. The tumor may grow outside the trachea or spread along alveolar walls, frequently forming nodules or masses 2-4 cm in diameter in the peripheral lung regions. Due to its rich vascular supply, adenocarcinoma tends to infiltrate locally and metastasize hematogenously early, often affecting the pleura and causing pleural effusion.
Large Cell Carcinoma (LCC)
LCC is an undifferentiated NSCLC, accounting for less than 10% of lung cancers. It lacks the cytological, histological, and immunophenotypic features of small cell carcinoma, adenocarcinoma, or squamous cell carcinoma. Diagnosis requires a surgical specimen and is not applicable to small biopsy or cytology samples. Immunohistochemistry and mucin staining are negative for squamous or glandular differentiation markers. LCC tends to metastasize later, making surgical resection a viable option.
Other Types
These include adenosquamous carcinoma, sarcomatoid carcinoma, NUT carcinoma (associated with nuclear protein of the testis gene rearrangement), and salivary gland carcinomas (e.g., adenoid cystic carcinoma, mucoepidermoid carcinoma).
Small Cell Lung Cancer (SCLC)
Lung neuroendocrine tumors include typical carcinoid, atypical carcinoid, small cell carcinoma, and large cell neuroendocrine carcinoma. SCLC is a poorly differentiated neuroendocrine tumor, encompassing small cell carcinoma and combined small cell carcinoma.
Cells are small, round or oval, with scant cytoplasm and indistinct cell borders. Nuclei are finely granular or hyperchromatic, with inconspicuous nucleoli and frequent mitotic figures.
Tumor cells contain neuroendocrine granules and secrete substances such as serotonin, catecholamines, histamine, and kinins, potentially causing carcinoid syndrome.
Tumor cells often express neuroendocrine markers such as CD56, NCAM, synaptophysin, and chromogranin. Ki-67 immunostaining is highly useful for distinguishing SCLC from carcinoid tumors, with SCLC showing a Ki-67 proliferation index of 50%-100%.
SCLC is characterized by rapid proliferation and early widespread metastasis. At diagnosis, 60%-88% of patients already have metastases to the brain, liver, bones, or adrenal glands, with only about 1/3 of cases confined to the thoracic cavity. SCLC is typically central in location, presenting as hilar masses and enlarged mediastinal lymph nodes causing cough and dyspnea. SCLC is highly sensitive to chemotherapy and radiotherapy. Among epithelial-origin lung cancers, squamous cell carcinoma, adenocarcinoma, large cell carcinoma, and small cell carcinoma account for approximately 90% of all cases.
Clinical Manifestations
The clinical manifestations of lung cancer are closely related to the tumor's size, type, stage of progression, location, and the presence of complications or metastases. Between 5% and 15% of patients are asymptomatic, with the disease being discovered during routine physical examinations or chest imaging studies. The remaining patients exhibit varying degrees of symptoms and signs associated with lung cancer.
Symptoms and Signs Caused by the Primary Tumor
Cough is an early symptom, often presenting as a dry, non-productive, and irritating cough. When the tumor causes bronchial narrowing, the cough may worsen and become persistent, with a high-pitched metallic quality or spasmodic choking. Mucinous adenocarcinoma may produce large amounts of mucus in the sputum. Secondary infections can lead to increased sputum production, which may become mucopurulent.
Hemoptysis or blood-streaked sputum is common in central lung cancer. Tumors growing into the bronchial lumen may cause intermittent or persistent blood-streaked sputum. Severe surface erosion and invasion of large blood vessels can result in massive hemoptysis.
Tumor growth into the trachea or bronchi can partially obstruct the airway. Metastasis to hilar lymph nodes may compress the main bronchus or carina. Other causes include pleural effusion, pericardial effusion, diaphragmatic paralysis, superior vena cava obstruction, or extensive lung involvement, leading to dyspnea, shortness of breath, and wheezing. Occasionally, stridor may occur. Auscultation may reveal localized or unilateral wheezing.
Dull chest pain may occur and is not necessarily related to tumor metastasis or direct invasion of the chest wall.
Tumor necrosis can cause fever. Most fever cases are due to obstructive pneumonia caused by the tumor, which often responds poorly to antibiotics.
Weight loss is a common manifestation of malignancy. In advanced stages, tumor-related toxins, infections, and pain can lead to reduced appetite, weight loss, or cachexia.
Symptoms and Signs Caused by Local Tumor Invasion
Tumor invasion of the pleura or chest wall can cause irregular, dull, or sharp pain, which worsens with breathing or coughing. Rib or spinal invasion may result in localized tenderness. Tumor compression of intercostal nerves may cause chest pain radiating to their distribution areas.
Tumor invasion or metastasis to mediastinal lymph nodes (commonly on the left side) can compress the recurrent laryngeal nerve, causing vocal cord paralysis and hoarseness.
Tumor invasion or compression of the esophagus can cause difficulty swallowing. It may also lead to tracheoesophageal fistulas, resulting in mediastinal or pulmonary infections.
Tumor metastasis to the pleura or obstruction of pulmonary lymphatic drainage can cause pleural effusion.
Tumor invasion of the pericardium or obstruction of cardiac lymphatic drainage may lead to pericardial effusion. Rapid or large pericardial effusions can cause cardiac tamponade symptoms.
Tumor invasion of the mediastinum, metastatic lymph node compression, or tumor thrombus in the superior vena cava can obstruct venous return. Symptoms include edema of the upper limbs, neck, and face, as well as dilated chest wall veins. Severe cases may exhibit cyanosis, conjunctival congestion, blurred vision, dizziness, and headaches.
Pancoast tumors (superior sulcus tumors) at the lung apex can compress the cervical sympathetic nerves, causing ipsilateral ptosis, miosis, enophthalmos, and anhidrosis or hypohidrosis of the forehead and chest wall. This is known as Horner syndrome.
Symptoms and Signs Caused by Distant Metastases
Pathological studies show that over 50% of squamous cell carcinoma cases, 80% of adenocarcinoma and large cell carcinoma cases, and more than 95% of small cell carcinoma cases involve extrapulmonary metastases. About 1/3 of symptomatic patients present with symptoms caused by distant metastases. Lung cancer can metastasize to any organ, causing corresponding symptoms and signs.
Brain metastases can cause symptoms of increased intracranial pressure, such as headaches, nausea, and emesis. Other symptoms include dizziness, ataxia, diplopia, personality changes, seizures, or unilateral limb weakness, which may progress to hemiplegia. Spinal cord compression can result in back pain, lower limb weakness, sensory abnormalities, and loss of bladder or bowel control.
Bone metastases cause localized pain and tenderness, and pathological fractures may occur. Commonly affected sites include ribs, spine, pelvis, and long bones of the limbs. Most cases exhibit osteolytic lesions.
Metastases to the liver, pancreas, and gastrointestinal tract can cause symptoms such as loss of appetite, right upper quadrant pain, jaundice, hepatomegaly, ascites, and symptoms of pancreatitis. Adrenal metastases are also common.
The supraclavicular lymph nodes are a common site of metastasis, often located in the posterior inferior region of the sternocleidomastoid muscle. These nodes are typically hard, fixed, and painless, gradually increasing in size and number, and may coalesce. Retroperitoneal lymph node metastases are also common.
Extrapulmonary Manifestations of Lung Cancer
Extrapulmonary manifestations of lung cancer that are not caused by metastases are referred to as paraneoplastic syndromes. These can occur before or after the diagnosis of lung cancer. Paraneoplastic syndromes are more common in small cell lung cancer (SCLC) and may present as initial symptoms or signs of recurrence. In some cases, the pathophysiology is clear (e.g., hormone secretion abnormalities), while in others, it remains unknown (e.g., anorexia, cachexia, weight loss, fever, and immunosuppression).
Tumor cells may secrete biologically active peptides and amines, such as adrenocorticotropic hormone (ACTH), parathyroid hormone (PTH), antidiuretic hormone (ADH), and gonadotropins, leading to corresponding clinical manifestations. Approximately 12% of lung cancer patients develop endocrine syndromes.
Syndrome of inappropriate antidiuretic hormone secretion (SIADH) presents as hyponatremia and hypo-osmolarity, with symptoms of water intoxication such as anorexia, nausea, emesis, drowsiness, irritability, disorientation, seizures, or coma. Hyponatremia may also result from increased secretion of ectopic atrial natriuretic peptide (ANP). Symptoms often resolve within 1-4 weeks of initiating chemotherapy.
Ectopic ACTH syndrome manifests as Cushing syndrome, including hyperpigmentation, edema, muscle atrophy, hypokalemia, metabolic alkalosis, hyperglycemia, or hypertension, though typical features like central obesity and purple striae are rare. This is commonly caused by SCLC or carcinoid tumors.
Mild cases present with thirst and polyuria, while severe cases may cause nausea, emesis, abdominal pain, constipation, drowsiness, or coma. Hypercalcemia is the most common life-threatening metabolic complication of malignancy and typically normalizes after tumor removal. It is most commonly seen in squamous cell carcinoma.
Ectopic gonadotropin secretion may cause mild gynecomastia in males, often accompanied by hypertrophic pulmonary osteoarthropathy, commonly seen in large cell carcinoma. Carcinoid syndrome, caused by excessive secretion of serotonin and other substances, is characterized by wheezing, flushing, watery diarrhea, and paroxysmal tachycardia, often seen in SCLC and adenocarcinoma.
Primary hypertrophic osteoarthropathy presents clubbed fingers (toes) in 30% of patients, most commonly in NSCLC. Affected bones may develop periostitis, causing pain, tenderness, and swelling, primarily in the distal long bones of the upper and lower limbs. X-rays show periosteal thickening and new bone formation, while bone scintigraphy reveals increased uptake at affected sites.
Paraneoplastic neurological syndrome and paraneoplastic myopathic syndrome are likely caused by autoimmune responses or tumor-secreted substances.
Eaton-Lambert myasthenic syndrome resembles myasthenia gravis, with proximal muscle weakness, especially in the pelvic girdle and lower limbs. Muscle strength temporarily improves with repeated activity. Reflexes may be diminished. Symptoms may improve with chemotherapy. Unlike myasthenia gravis, over 70% of patients do not respond well to neostigmine, and electrophysiological studies show decremental responses to low-frequency stimulation and incremental responses to high-frequency stimulation. Common in SCLC.
Lung cancer can cause polyneuropathy, subacute cerebellar degeneration, cortical degeneration, and polymyositis. SCLC is associated with paraneoplastic encephalomyelitis, sensory neuropathy, cerebellar degeneration, limbic encephalitis, and brainstem encephalitis, often accompanied by anti-neuronal antibodies such as anti-Hu, anti-CRMP5, and ANNA-3 antibodies.
1% - 8% of patients experience coagulation or thrombotic abnormalities, including migratory thrombophlebitis (Trousseau syndrome), nonbacterial thrombotic endocarditis with atrial thrombi, disseminated intravascular coagulation (DIC) with hemorrhage, anemia, leukocytosis, and leukoerythroblastosis. Thrombotic complications in lung cancer are associated with poor prognosis. Other conditions include dermatomyositis, acanthosis nigricans (1%), nephrotic syndrome, and glomerulonephritis (≤1%).
Imaging and Other Diagnostic Examinations
Imaging Examinations
Chest X-ray is one of the most commonly used methods for detecting lung cancer. However, its resolution is low, making it difficult to detect small pulmonary nodules or lesions in hidden areas, which limits its ability to identify early-stage lung cancer. Common radiographic features of lung cancer are as follows.
Central Lung Cancer
Tumors grow in the main bronchi, lobar bronchi, or segmental bronchi.
Tumor growth into the bronchial lumen can cause signs of bronchial obstruction. This often appears as a unilateral hilar round opacity with irregular or lobulated margins. When associated with atelectasis or obstructive pneumonia, the lower border may show Golden S sign, which is a typical feature of right upper lobe central lung cancer.
Tumor growth within the bronchus can partially or completely obstruct the bronchial lumen, leading to localized emphysema, atelectasis, obstructive pneumonia, or secondary lung abscesses.
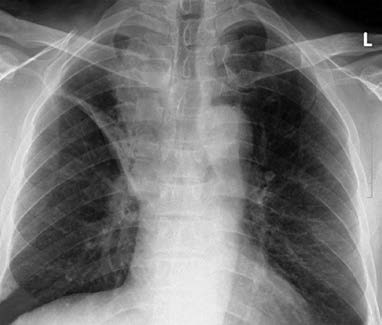
Figure 1 Central lung cancer
Male, 60 years old. Central lung cancer in the right upper lobe with obstructive atelectasis and obstructive pneumonia. Pathology: Pulmonary squamous cell carcinoma.
Peripheral Lung Cancer
Tumors originate in bronchi distal to the segmental bronchi. In the early stages, they often appear as localized, patchy opacities with ill-defined margins and low density. They may also present as nodular, spherical, reticular, or ground-glass opacities, which can be misdiagnosed as inflammation or tuberculosis. As the tumor grows, the opacity enlarges, becomes denser, and appears round or oval with lobulated margins, umbilication, or fine spiculations. Pleural traction is often observed. If the tumor metastasizes to hilar lymph nodes, thickened lymphatic vessels may appear as irregular linear opacities accompanied by enlarged hilar lymph nodes. When necrosis occurs in the tumor and communicates with the bronchus, it may present as a thick-walled, eccentric, irregularly contoured cavitary lesion. Secondary infections can result in fluid levels within the cavity. Adenocarcinoma spreading through the bronchi may mimic bronchopneumonia with patchy infiltrative opacities. Pleural invasion can cause pleural effusion, while rib invasion can lead to bone destruction.
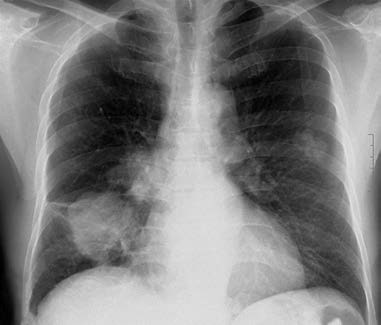
Figure 2 Peripheral lung cancer
Male, 52 years old. Peripheral lung cancer in the right lower lobe with right hilar lymph node involvement and bilateral lung metastases. Pathology: Well-differentiated pulmonary adenocarcinoma.
Chest CT has higher resolution and can detect small pulmonary lesions and areas not easily visible on standard chest X-rays (e.g., behind the heart, near the spine, in the lung apex, costophrenic angles, and rib heads). Contrast-enhanced CT is sensitive for detecting hilar and mediastinal lymphadenopathy, aiding in clinical staging of lung cancer. Spiral CT can reveal nodules smaller than 5 mm, central airway lesions, and the sixth to seventh generations of bronchial branches and small blood vessels, enabling a clear assessment of the relationship between the lesion and surrounding airways and blood vessels. Low-dose CT is effective for detecting early-stage lung cancer and has replaced chest X-ray as a more sensitive tool for evaluating pulmonary nodules. CT-guided percutaneous lung biopsy is an important histological diagnostic technique. CT simulation imaging can guide bronchoscopy for biopsies within the airway or through the bronchial wall.
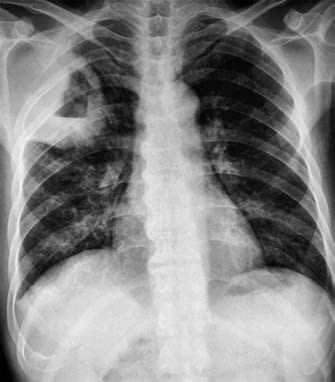
Figure 3 Cancerous cavitation
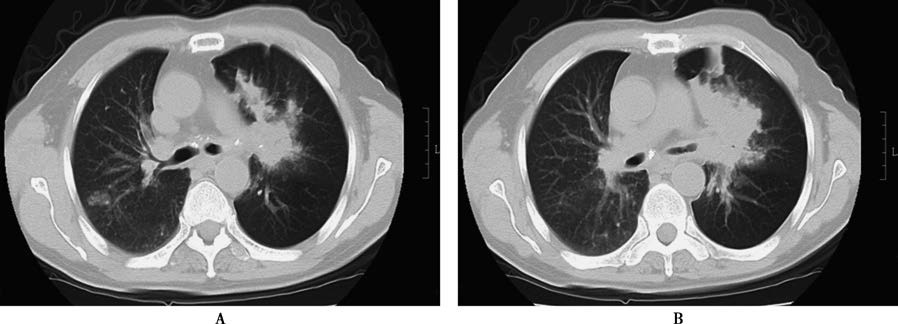
Figure 4 Small cell lung cancer
Male, 59 years old. Central lung cancer in the left lung involving the left main bronchus and upper and lower lobar bronchi, with obstructive pneumonia in the left lung and multiple metastatic lesions in the lung. Bronchoscopic biopsy pathology: Small cell lung cancer.
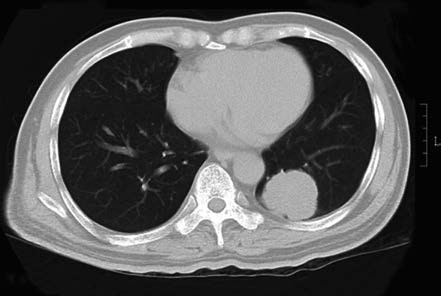
Figure 5 Left lower lobe adenocarcinoma
Male, 61 years old. Peripheral lung cancer in the left lower lobe. Pathology: Pulmonary adenocarcinoma.
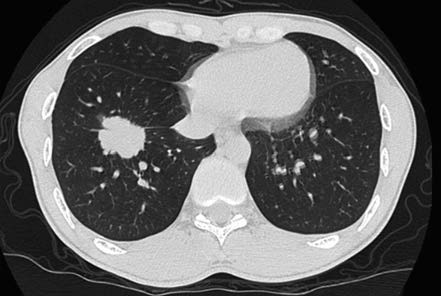
Figure 6 Right lower lobe adenocarcinoma
Male, 32 years old. Peripheral lung cancer in the right lower lobe. Pathology: Pulmonary adenocarcinoma.
Compared to CT, MRI is superior in assessing the relationship between tumors and large blood vessels and in detecting brain parenchymal or meningeal metastases. However, it is less sensitive than CT for detecting small pulmonary lesions (<5 mm).
Bone scintigraphy can assess the presence of bone metastases, with sensitivities, specificities, and accuracies of 91%, 88%, and 89%, respectively. Using radionuclide-labeled somatostatin analogs can further aid in staging small cell lung cancer (SCLC).
Positron emission tomography (PET) tracks radiolabeled compounds to show physiological changes in metabolism within the body, providing non-invasive visualization of tissue and organ function with quantitative analysis. PET-CT combines PET and CT, allowing for simultaneous acquisition of CT anatomical images and PET functional metabolic images through a rapid whole-body scan. This provides both metabolic information and precise anatomical localization, making it superior to other imaging modalities for detecting early-stage lung cancer, metastatic lesions, tumor staging, and treatment evaluation. However, it is important to note that positive findings on PET-CT still require cytological or pathological confirmation for a definitive diagnosis.
Pathological Diagnostic Examinations
Sputum cytology is the simplest and most convenient non-invasive diagnostic method for central lung cancer, but it carries a risk of false positives and false negatives. To improve the detection rate, deep airway sputum samples should be collected and promptly sent for examination, with at least three samples submitted. Sensitivity is less than 70%, and subtyping is challenging, but specificity is high.
Thoracentesis allows for the collection of pleural effusion for cytological examination, with a detection rate of 40%-90%. This helps confirm pathology and stage lung cancer. Centrifuged cell blocks from pleural effusion can be embedded in paraffin, sectioned, and stained to improve the positive diagnostic rate. Similar procedures can be applied to metastatic serous effusions in other locations.
Bronchoscopy is one of the main methods for diagnosing lung cancer. It can access the fourth to fifth generations of bronchial branches, allowing direct visualization of approximately 1/3 of the proximal bronchial mucosa. Tissue or cytological samples can be obtained through biopsy, brushing, or lavage. Combining these techniques increases the detection rate. Fluorescence bronchoscopy, which utilizes the autofluorescence properties of tumor tissue, can significantly improve the detection of epithelial dysplasia and invasive lung cancer when combined with conventional bronchoscopy.
For lesions not visible with conventional bronchoscopy, advanced techniques such as thin or ultrathin bronchoscopy, X-ray fluoroscopy, radial ultrasound probes, or electromagnetic navigation bronchoscopy can guide biopsies based on the lesion's location and available resources. Endobronchial ultrasound (EBUS)-guided transbronchial needle aspiration (TBNA) aids in diagnosing airway wall infiltration, extraluminal lesions, and mediastinal lymph node involvement, as well as in TNM staging of lung cancer. Peripheral lesions can be biopsied using small ultrasound probes.
Thoracoscopy is used for subpleural lesions when bronchoscopy and other methods fail to obtain pathological samples. It also allows for the assessment of pleural metastases, providing reliable information for comprehensive treatment planning.
Mediastinoscopy is effective for differentiating benign from malignant diseases associated with mediastinal lymphadenopathy. It is also a valuable method for staging lung cancer and preoperative lymph node assessment, though it is relatively invasive and carries higher risks.
Needle aspiration or cutting biopsies of lesions can be performed under X-ray fluoroscopy, chest CT, or ultrasound guidance. This minimally invasive procedure is simple, quick, and suitable for lesions close to or adjacent to the chest wall.
Enlarged superficial lymph nodes in the supraclavicular or axillary regions can be biopsied via fine-needle aspiration or surgical excision. This procedure is simple and can be performed on an outpatient basis.
If the above examinations fail to provide a definitive diagnosis, open lung biopsy may be considered. The decision should carefully weigh the risks and benefits based on the patient's age, pulmonary function, and other factors.
Tumor Marker Testing
Currently, no tumor markers have high sensitivity and specificity for diagnosing lung cancer. However, markers such as carcinoembryonic antigen (CEA), neuron-specific enolase (NSE), cytokeratin 19 fragment antigen 21-1 (CYFRA21-1), and pro-gastrin-releasing peptide (ProGRP) may have reference value for diagnosis and monitoring disease progression.
Lung cancer is believed to result from the activation of oncogenes and the loss of tumor suppressor genes. The detection of oncogene products, such as MYC gene amplification, RAS gene mutations, and abnormalities in tumor suppressor genes like RB1 and TP53, can aid in diagnosing early-stage lung cancer. Genetic testing can also identify patients who may benefit from targeted therapies.
Current testing focuses on detecting EGFR mutations, ALK fusion genes, ROS1 rearrangements, BRAF V600 mutations, RET rearrangements, MET exon 14 skipping mutations, and NTRK1/2/3 rearrangements in non-small cell lung cancer (NSCLC) patients. Expanded testing includes MET amplification or overexpression and HER-2. Resistance mutations, such as EGFR T790M and C797S or MET amplification, can also be detected.
When tumor tissue samples are difficult to obtain, circulating tumor DNA (ctDNA) from peripheral blood can be used to assess gene mutation status, a method known as liquid biopsy. Immunohistochemical testing for PD-L1 expression can help identify NSCLC patients who may benefit from immune checkpoint inhibitors. Tumor mutation burden (TMB) may also predict the efficacy of immunotherapy, though there is currently no standardized method or threshold for its evaluation.
Diagnosis
The diagnosis of lung cancer can be conducted following these steps:
Localization by CT
For patients with clinical symptoms or radiological signs suggestive of lung cancer, chest and abdominal CT scans should be performed first. This helps identify the primary tumor site, mediastinal lymph node involvement, and the extent of spread to other anatomical regions.
Histopathological Diagnosis
Patients suspected of having lung cancer must undergo histological sampling for diagnosis. Tumor tissue can often be obtained through minimally invasive techniques such as bronchoscopy or thoracoscopy. However, single sputum cytology is not recommended for definitive diagnosis. Biopsies should also be performed on palpable superficial lymph nodes or skin metastases. If distant metastases are suspected, tissue samples should be obtained from soft tissue masses, lytic bone lesions, bone marrow, pleural, or liver lesions. For pleural effusion, sufficient cell clusters should be collected, or thoracoscopy should be performed. For patients highly suspected of having stage I or II lung cancer, direct surgical resection is currently recommended.
Molecular Pathological Diagnosis
If conditions allow, molecular testing should be conducted simultaneously with pathological confirmation to detect mutations in tumor tissue, including EGFR mutations, ALK fusion genes, ROS1 fusion genes, BRAF V600, RET, MET exon 14 skipping mutations, KRAS, and NTRK. For non-small cell lung cancer (NSCLC), PD-L1 expression levels may also be considered to facilitate the development of personalized treatment plans.
Differential Diagnosis
Lung cancer often coexists with or mimics certain pulmonary diseases in imaging, leading to misdiagnosis or missed diagnosis. Clinicians should differentiate lung cancer from the following conditions:
Tuberculosis
Pulmonary tuberculoma is common in young patients and often asymptomatic. Lesions are typically located in the apical-posterior segment of the upper lobe or the dorsal segment of the lower lobe. They appear as well-defined, hyperdense nodules with a capsule and sometimes calcifications. Surrounding fibronodular lesions may be present, and the lesion remains unchanged for years.
Hilar lymph node tuberculosis can be confused with central lung cancer. It is more common in children and young adults and is associated with symptoms of tuberculosis, such as fever and diaphoresis. Tuberculin skin tests are often positive, and the condition responds well to anti-tuberculosis treatment.
Acute miliary tuberculosis can be seen in younger patients with systemic symptoms like fever and diaphoresis. On X-ray, it appears as fine, homogeneous, hypodense, miliary nodules. Adenocarcinoma (formerly known as bronchioloalveolar carcinoma) may present with multiple nodular lesions of varying sizes in both lungs, with clear margins, higher density, and progressive growth.
Pneumonia
Pneumonia is associated with symptoms such as fever, cough, and expectoration and typically responds well to antibiotic treatment. If no systemic symptoms are present, but pulmonary opacities resolve slowly after antibiotic therapy or recurrent pneumonia occurs in the same location, lung cancer should be considered. Chronic inflammatory consolidation forming a mass-like inflammatory pseudotumor may also mimic lung cancer. However, inflammatory pseudotumors often have irregular shapes, uneven margins, higher core density, pleural thickening, and remain unchanged over time.
Lung Abscess
Lung abscess has an acute onset with severe systemic symptoms, including chills, high fever, cough, and production of large amounts of foul-smelling purulent sputum. Imaging typically shows homogeneous patchy opacities with fluid levels in cavities. In contrast, cancerous cavities usually do not cause fever but may present with clinical features of a lung abscess if secondary infection occurs. Imaging of cancerous cavities shows eccentric cavities with thick, irregular inner walls. Bronchoscopy and sputum cytology can help differentiate the two conditions.
Tuberculous Pleurisy
Tuberculous pleurisy should be differentiated from malignant pleural effusion.
Pulmonary Cryptococcosis
Pulmonary cryptococcosis may present as solitary or multiple nodules and masses, often located subpleurally. Solitary lesions can be confused with peripheral lung cancer. Lung biopsy and serum cryptococcal capsular polysaccharide antigen testing can aid in differentiation.
Other Conditions
Other conditions, such as benign lung tumors and lymphoma, require differentiation through histopathological examination.
Clinical Staging
In 2023, the International Association for the Study of Lung Cancer (IASLC) published the 9th edition of the revised TNM staging system for lung cancer.
For small cell lung cancer (SCLC), staging can also be divided into limited-stage SCLC and extensive-stage SCLC:
- Limited-stage SCLC: Refers to lesions confined to one hemithorax that can be safely encompassed within a single radiation field.
- Extensive-stage SCLC: Refers to lesions extending beyond one hemithorax, including malignant pleural or pericardial effusion and hematogenous metastases.
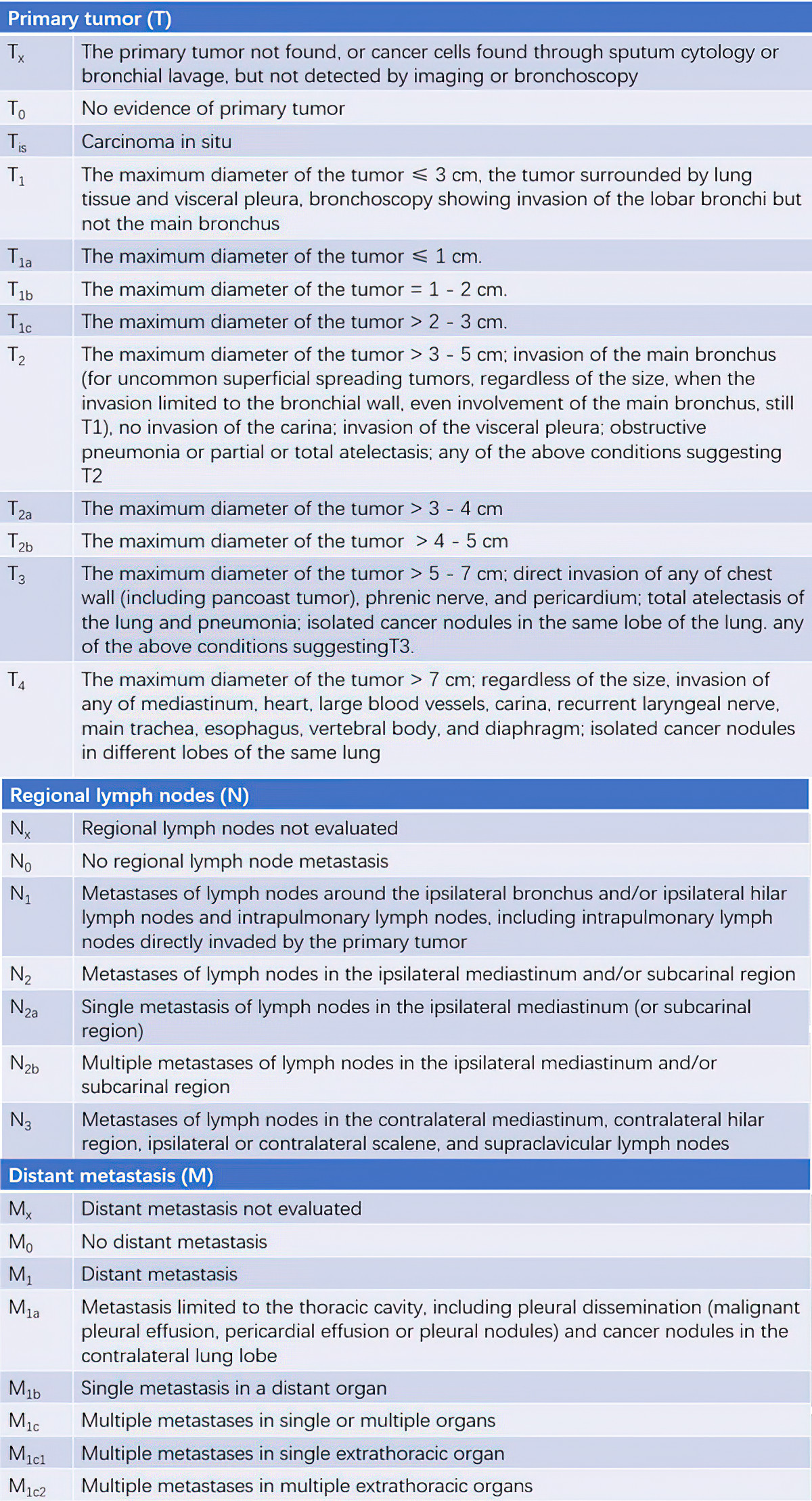
Table 1 TNM staging for lung cancer
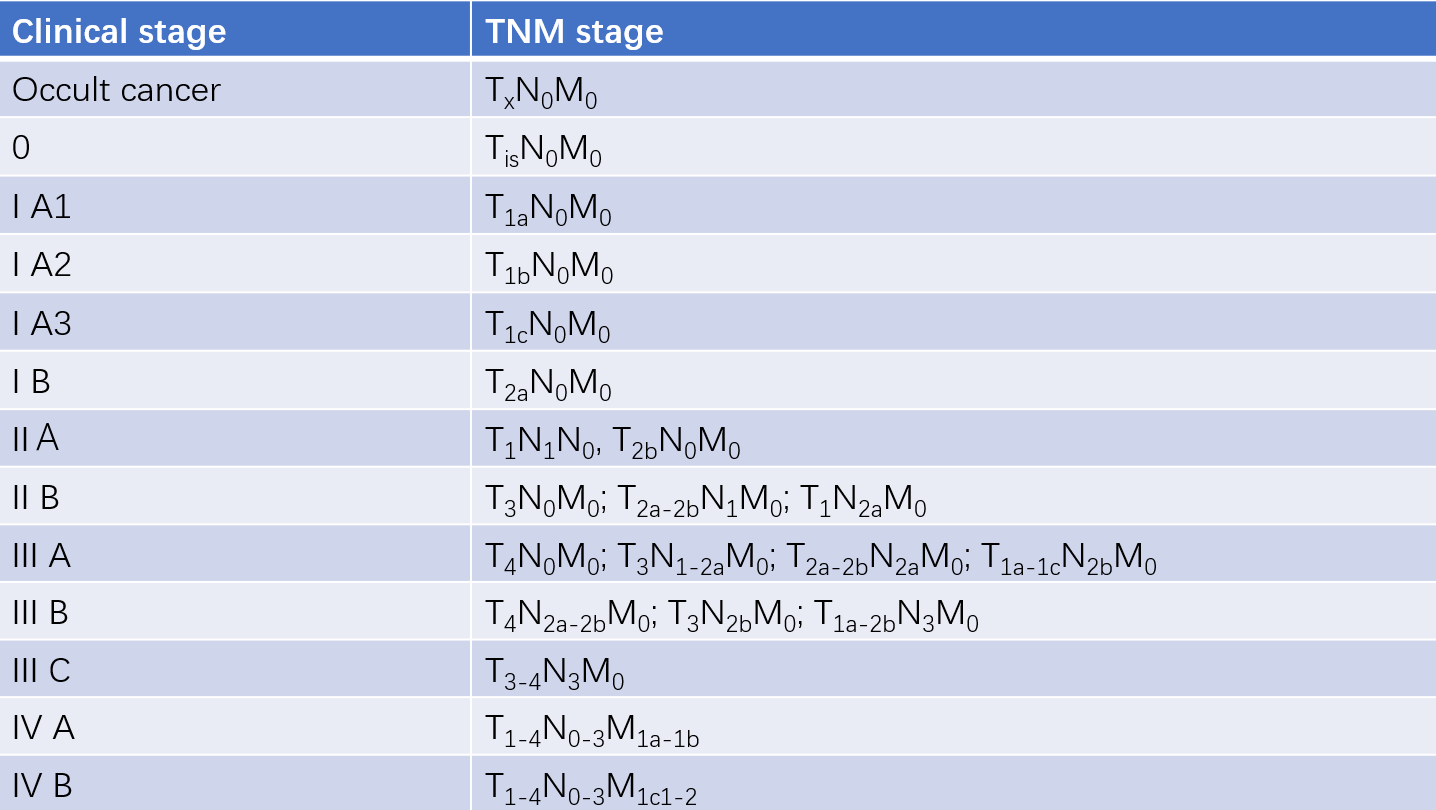
Table 2 Relationship between TNM staging and clinical staging
Treatment
The treatment of lung cancer should be based on the patient's performance status (PS) score, pathological type (including molecular pathology diagnosis), and extent of disease (clinical staging). A multidisciplinary and comprehensive approach is emphasized, with a focus on personalized treatment. The goal is to rationally and systematically apply surgery, chemotherapy, targeted therapy, immunotherapy, and radiotherapy to achieve either curative outcomes or maximum tumor control, thereby improving cure rates, enhancing quality of life, and prolonging survival.

Table 3 Performance status (PS) score
Surgical Treatment
Surgery is the best treatment for early-stage lung cancer. It can be classified as curative or palliative. Efforts should be made to achieve curative resection, aiming to remove the tumor, reduce metastasis and recurrence, and allow for TNM staging to guide postoperative comprehensive treatment.
NSCLC
Surgery is primarily suitable for stage I and II patients, with curative resection being the first-line treatment. For patients with stage IIIa (T3N1, T4N0-1, and some T1-3N2), a multidisciplinary approach is required, including surgery combined with postoperative chemotherapy, sequential chemoradiotherapy, or concurrent chemoradiotherapy. For stage II-III NSCLC, adjuvant chemotherapy is required after curative surgery. Neoadjuvant chemotherapy (preoperative chemotherapy) can downstage previously inoperable tumors to make surgery feasible. Postoperative treatments, based on the final pathological TNM stage and surgical margin status, may include reoperation, adjuvant targeted therapy, chemotherapy, immunotherapy, or radiotherapy. For patients who cannot tolerate lobectomy, wedge resection may be considered.
SCLC
Over 90% of patients present with intrathoracic or distant metastases at diagnosis, making surgery generally unsuitable. However, for T1-2N0 patients confirmed by pathological mediastinal staging (e.g., mediastinoscopy or mediastinotomy), lobectomy with lymph node dissection can be considered. Since surgery alone cannot cure SCLC, all postoperative SCLC patients should receive 4-6 cycles of platinum-based chemotherapy.
Pharmacological Treatment
Pharmacological treatment primarily includes chemotherapy, immunotherapy, and targeted therapy/anti-angiogenic therapy, which are mainly used for advanced or recurrent lung cancer. Chemotherapy and immunotherapy can also be used as adjuvant treatments after surgery, as neoadjuvant therapy before surgery, or in combination with radiotherapy.
Chemotherapy should be carefully tailored to the patient's disease stage, performance status, personal preferences, drug side effects, and quality of life, avoiding both overtreatment and undertreatment. Patients with a PS score ≤2 and adequate organ function may receive chemotherapy. Commonly used drugs include platinum-based agents (cisplatin, carboplatin), gemcitabine, pemetrexed, taxanes (paclitaxel, docetaxel), vinorelbine, etoposide, and camptothecin analogs (irinotecan).
- First-line chemotherapy: A platinum-based doublet regimen is recommended.
- Second-line chemotherapy: Docetaxel or pemetrexed monotherapy is recommended.
Treatment efficacy is typically evaluated in two cycles, with close monitoring and management of adverse effects, and dosage adjustments as needed.
Targeted therapy focuses on specific molecular abnormalities in tumor tissues or cells, such as driver gene mutations and tumor-related signaling pathways. By using molecularly targeted drugs to block these pathways, targeted therapy selectively reverses the malignant biological behavior of tumor cells at the molecular level, inhibiting tumor growth or even causing tumor regression.
- EGFR mutations: EGFR-TKIs such as erlotinib, gefitinib, afatinib, osimertinib, and almonertinib.
- ALK rearrangements: ALK inhibitors such as crizotinib, alectinib, ceritinib, lorlatinib, and ensartinib.
- ROS1 rearrangements: Crizotinib or entrectinib.
- MET exon 14 skipping mutations: Capmatinib.
- BRAF V600 mutations: Dabrafenib combined with trametinib.
- RET fusions: Selpercatinib.
Targeted therapy is mainly used for adenocarcinoma patients with NSCLC who are not suitable for curative treatment or have advanced or metastatic disease. It can significantly prolong survival. The success of targeted therapy depends on selecting specific patient populations with the corresponding molecular targets.
Tumor angiogenesis is one of the hallmarks of malignancy. The VEGF-VEGFR2 signaling pathway controls endothelial cell proliferation, survival, and migration, ultimately promoting new blood vessel formation. Tumor growth, progression, and metastasis rely on angiogenesis, and anti-VEGF therapy can effectively inhibit tumor growth and metastasis.
Anti-angiogenic agents include:
- VEGF monoclonal antibody (bevacizumab).
- VEGFR monoclonal antibody (ramucirumab).
- VEGFR tyrosine kinase inhibitors (sorafenib, anlotinib, nintedanib, apatinib, regorafenib).
Recent studies suggest that combining anti-angiogenic therapy with chemotherapy, targeted therapy, or immunotherapy yields significant benefits for advanced lung cancer. For example, bevacizumab combined with chemotherapy improves chemotherapy efficacy and extends progression-free survival in advanced NSCLC. Anlotinib, a novel small-molecule multi-target tyrosine kinase inhibitor, has dual effects of anti-angiogenesis and tumor growth inhibition. It has shown significant benefits in third-line or later treatment of advanced NSCLC, extending progression-free survival and overall survival.
Immune checkpoint inhibitors (ICIs) targeting PD-1/PD-L1 can block the interaction between PD-1 and PD-L1 on tumor cells, activating anti-tumor immune responses. ICIs have revolutionized the treatment of advanced NSCLC without driver mutations, increasing the 5-year survival rate from 5% in the chemotherapy era to 13.4%-23.2%. ICIs are now the standard treatment for advanced NSCLC.
Common ICIs include pembrolizumab, atezolizumab, camrelizumab, sintilimab, tislelizumab, sugemalimab, toripalimab, and durvalumab.
NSCLC
Chemotherapy is less effective for NSCLC. For advanced and recurrent NSCLC, combination chemotherapy can alleviate symptoms, improve quality of life, and increase survival rates, with partial response rates of 30%-40%, complete response rates of 5%, median survival of 9-10 months, and a 1-year survival rate of 30%-40%.
- First-line chemotherapy: Platinum-based doublet regimens, such as carboplatin or cisplatin combined with paclitaxel, vinorelbine, gemcitabine, pemetrexed, or docetaxel, for 4-6 cycles.
- Maintenance therapy: For patients whose tumors respond or remain stable after chemotherapy without progression.
- Targeted therapy: EGFR-TKIs (e.g., osimertinib, almonertinib) are superior to chemotherapy for EGFR-mutant stage IV NSCLC in terms of response rate and progression-free survival (PFS), with fewer side effects. Resistance mutations (e.g., T790M) can be treated with osimertinib or almonertinib.
For ALK or ROS1 rearrangements, ALK inhibitors such as crizotinib or ceritinib are recommended.
For non-squamous NSCLC without hemoptysis or brain metastases, anti-angiogenic agents like bevacizumab can be combined with chemotherapy.
For PD-L1-positive (≥1%) NSCLC without driver mutations, first-line chemotherapy combined with immunotherapy is recommended. For PD-L1-high expression (≥50%), ICIs alone may provide significant benefits.
SCLC
SCLC is highly sensitive to chemotherapy, which forms the backbone of treatment. First-line regimens include etoposide or irinotecan combined with cisplatin or carboplatin for 4-6 cycles.
For limited-stage SCLC (stage II-III), combined chemoradiotherapy is recommended.
For extensive-stage SCLC, combination chemotherapy and immunotherapy are the mainstay treatments, with common regimens including etoposide + carboplatin + atezolizumab, etoposide + platinum + durvalumab, or etoposide + platinum + adebrelimab.
For extensive-stage SCLC with brain metastases, chemotherapy may be given before or after whole-brain radiotherapy, depending on neurological symptoms. Most limited-stage and nearly all extensive-stage SCLC cases will eventually relapse. For relapsed SCLC, second-line chemotherapy or reusing first-line regimens may be considered based on the type of relapse.
Radiation Therapy
Radiation therapy can be categorized into curative radiation therapy, palliative radiation therapy, adjuvant radiation therapy, neoadjuvant chemoradiotherapy, and prophylactic radiation therapy.
Curative radiation therapy is used for localized lesions in patients who are not suitable for surgery due to anatomical or other reasons. When combined with chemotherapy, its efficacy can be enhanced.
Palliative radiation therapy aims to suppress tumor progression, delay tumor spread, and alleviate symptoms. It is effective for symptoms caused by lung cancer, such as persistent cough, hemoptysis, atelectasis, and superior vena cava syndrome. It can also relieve pain from bone metastases and symptoms caused by brain metastases.
Adjuvant radiation therapy is suitable for patients undergoing preoperative radiation therapy or those with positive surgical margins postoperatively.
Prophylactic radiation therapy is used for small cell lung cancer (SCLC) patients who have responded to systemic treatment, particularly for whole-brain radiation therapy.
Radiation therapy is usually combined with chemotherapy to treat lung cancer. Depending on the stage, treatment goals, and the patient's overall condition, combination approaches such as concurrent chemoradiotherapy or sequential chemoradiotherapy may be chosen. Patients undergoing chemoradiotherapy are at higher risk of infection, requiring careful protection of the lungs, heart, esophagus, and spinal cord. Efforts should be made to avoid unplanned interruptions in radiation therapy due to improper management of side effects.
The sensitivity of lung cancer to radiation therapy is highest in SCLC, followed by squamous cell carcinoma and adenocarcinoma. Therefore, the radiation dose is lowest for SCLC and highest for adenocarcinoma. Typical doses range from 40-70 Gy over 5-7 weeks. Common radiation modalities include cobalt-60 gamma rays, electron beam beta rays, and neutron accelerators. Efforts should be made to minimize and prevent adverse effects such as leukopenia, radiation pneumonitis, and radiation esophagitis. Radiation therapy is contraindicated in patients with severe systemic conditions, including significant dysfunction of the heart, lungs, liver, or kidneys. Advanced and safer techniques, such as three-dimensional conformal radiation therapy (3DCRT) and intensity-modulated radiation therapy (IMRT), should be reasonably utilized.
NSCLC radiation therapy applications:
- Locally advanced patients, in combination with chemotherapy.
- Curative treatment for early-stage NSCLC patients who cannot undergo surgery due to physical conditions.
- Selective preoperative or postoperative adjuvant therapy.
- Treatment of local recurrence or metastases.
- Palliative treatment for advanced, incurable patients.
SCLC radiation therapy applications:
- For limited-stage SCLC, after systemic chemotherapy, some patients achieve complete remission but have a high risk of intrathoracic recurrence and brain metastases. Adding thoracic radiation therapy and prophylactic cranial irradiation can significantly reduce the rates of local recurrence and brain metastases, as well as mortality risk.
- For extensive-stage SCLC, after distant metastases are controlled with chemotherapy, adding thoracic radiation therapy can improve tumor control and prolong survival.
Interventional Therapy
Bronchial artery infusion chemotherapy is suitable for advanced-stage patients who have lost indications for surgery and are unresponsive to systemic chemotherapy. It has minimal side effects, alleviates symptoms, and reduces patient suffering.
Bronchoscopic interventional therapy:
- Photodynamic laser therapy and YAG laser resection: These techniques can remove intraluminal tumors in the airway, relieve airway obstruction, and control bleeding, thereby prolonging survival.
- Endobronchial brachytherapy: This can alleviate symptoms such as obstruction and hemoptysis caused by tumors.
- Ultrasound-guided interventions: Anti-tumor drugs and other agents can be directly injected into the tumor tissue.
Prevention
Lung cancer prevention involves three levels of preventive measures:
Primary Prevention
This involves addressing the root causes of cancer and enhancing the body’s ability to prevent cancer, aiming to stop the disease before it occurs. Measures include eliminating carcinogenic and cancer-promoting factors, such as quitting smoking, reducing air pollution, maintaining a healthy diet, avoiding overwork, improving harmful environments (both living and occupational), and changing unhealthy lifestyle habits. By eliminating or reducing exposure to carcinogens and cancer-promoting factors, the risk of cancer can be significantly reduced.
Secondary Prevention
This refers to preclinical prevention. The effectiveness and prognosis of lung cancer treatment depend on early diagnosis and treatment, making the early detection of high-risk populations particularly important. Screening programs for high-risk individuals are critical. Over the years, domestic and international efforts have focused on lung cancer screening to achieve early diagnosis and treatment, ultimately reducing lung cancer-related mortality.
A 2011 randomized controlled trial conducted by the U.S. National Lung Screening Trial (NLST) demonstrated that, compared to chest X-rays, low-dose computed tomography (LDCT) screening for high-risk individuals reduced lung cancer mortality by 20%. Screening guidelines from several authoritative medical organizations in Europe and the U.S. recommend LDCT screening for high-risk populations.
Tertiary Prevention
This refers to clinical treatment or rehabilitative prevention. The goal is to reduce complications, prevent disease progression, and avoid disability in cancer patients, thereby improving their quality of life.
These three levels of prevention are interconnected and collectively form a comprehensive strategy for preventing lung cancer.
Prognosis
The prognosis of lung cancer depends on early detection, diagnosis, and treatment. Due to insufficient early diagnosis, the prognosis for lung cancer remains poor, with approximately 80% of patients dying within five years after diagnosis. Only 15% of patients are diagnosed with localized disease, for whom the five-year survival rate can reach 50%.