Definition
Hemolysis refers to the destruction of red blood cells (RBCs) and the shortening of their lifespan. The bone marrow has a compensatory capacity of producing RBCs at 6 to 8 times the normal hematopoietic rate. Hemolytic anemia (HA) occurs when the degree of hemolysis exceeds the bone marrow's compensatory capacity, leading to anemia. When hemolysis occurs but the bone marrow is able to compensate, there may be no anemia, a condition referred to as a hemolytic state. HA accounts for approximately 5% of all cases of anemia and can occur at any age.
Clinical Classification
Hemolytic anemia can be classified in various ways. According to onset and severity, it can be divided into acute hemolysis and chronic hemolysis (as detailed under clinical manifestations). Based on the site of hemolysis, it can be classified as intravascular hemolysis and extravascular hemolysis (as detailed under pathogenesis). According to etiology, it can be categorized into HA caused by intrinsic abnormalities of RBCs and HA caused by extrinsic factors, as outlined below:
HA Caused by Intrinsic Abnormalities of RBCs
RBC Membrane Abnormalities
Inherited RBC membrane abnormalities includes hereditary spherocytosis, hereditary elliptocytosis, hereditary acanthocytosis, and hereditary stomatocytosis.
Acquired abnormalities in GPI-anchored membrane proteins includes paroxysmal nocturnal hemoglobinuria (PNH).
Inherited RBC Enzyme Deficiencies
Enzyme deficiencies in the pentose phosphate pathway includes glucose-6-phosphate dehydrogenase (G-6-PD) deficiency.
Enzyme deficiencies in the anaerobic glycolysis pathway includes pyruvate kinase deficiency.
Additional enzyme deficiencies, such as those involving nucleotide metabolism or oxidoreductase pathways, can also lead to HA.
Inherited Disorders of Globin Synthesis
Structural abnormalities in globin chains includes hemoglobinopathies.
Quantitative abnormalities in globin chain production includes thalassemias, also known as thalassemia syndromes.
HA Caused by Extrinsic Factors
Immune-Related HA
Autoimmune HA includes HA due to warm antibodies or cold antibodies (e.g., cold agglutinin type or Donath-Landsteiner type). This category includes primary cases or those secondary to conditions such as systemic lupus erythematosus (SLE), viral infections, or drug-induced causes.
Alloimmune HA includes conditions such as hemolytic reactions due to transfusion of incompatible blood and hemolytic disease of the newborn.
Vascular-Related HA
Microangiopathic HA includes thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS), disseminated intravascular coagulation (DIC), and sepsis.
Valve-related HA includes hemolysis caused by conditions such as calcific aortic valve stenosis and prosthetic heart valves.
Repetitive mechanical trauma to the vessel wall includes conditions such as march hemoglobinuria.
Biological Factors
These includes causes such as snake venom, malaria, and kala-azar (visceral leishmaniasis).
Physical and Chemical Factors
Extensive burns, changes in plasma osmotic pressure, and chemical agents (e.g., phenylhydrazine, nitrites) can induce hemolytic anemia through mechanisms such as acquired methemoglobinemia.
Pathogenesis
The mechanisms of RBC destruction differ depending on the underlying cause of hemolytic anemia (HA) (detailed in specific subsections). The destruction of RBCs occurs either intravascularly or extravascularly, leading to corresponding clinical manifestations and laboratory findings. Additionally, destruction of immature erythroblasts within the bone marrow, known as ineffective erythropoiesis or in situ hemolysis, occurs before their release into the bloodstream. This process, essentially a form of extravascular hemolysis, is commonly observed in conditions such as megaloblastic anemia.
Increased Destruction of RBCs
Intravascular Hemolysis
Intravascular hemolysis involves the destruction of RBCs within the bloodstream, releasing free hemoglobin, which leads to hemoglobinemia. Free hemoglobin immediately binds with plasma haptoglobin to form a complex, which is transported to the liver for further processing. In the liver, heme from hemoglobin is metabolized into iron and biliverdin, with biliverdin subsequently converted into bilirubin.
In cases of pronounced intravascular hemolysis that overwhelm haptoglobin-binding capacity, free hemoglobin is filtered through the glomeruli. When the amount exceeds the proximal tubules' reabsorptive capacity, hemoglobinuria occurs, indicating rapid intravascular hemolysis. Free hemoglobin reabsorbed by renal proximal tubular epithelial cells is further broken down into porphyrin, globin, and iron. Iron is deposited within these epithelial cells in the form of ferritin or hemosiderin, which is eventually shed into the urine as hemosiderin. This indicates chronic intravascular hemolysis.
Extravascular Hemolysis
Extravascular hemolysis involves the destruction of RBCs by the mononuclear phagocyte system, primarily in the spleen. Hemoglobin released from these RBCs is broken down into globin and heme, with heme further degraded into bilirubin.
Regardless of the location of hemoglobin breakdown, bilirubin is one of its end products. Unconjugated bilirubin is taken up by hepatocytes, conjugated with glucuronic acid to form conjugated bilirubin, and excreted into the bile. In the intestine, conjugated bilirubin is reduced by intestinal bacteria to stercobilinogen and excreted in the feces.
A small portion of stercobilinogen is reabsorbed into the bloodstream via the intestines and re-secreted into the bile by hepatocytes, forming the enterohepatic circulation of stercobilinogen. A minor fraction enters the kidneys and is excreted in the urine as urobilinogen. When the hemolysis rate exceeds the liver's bilirubin-processing capacity, hemolytic jaundice may occur. Chronic extravascular hemolysis, due to prolonged hyperbilirubinemia, can result in hepatocellular damage and elevated conjugated bilirubin levels.
Compensatory Erythropoietic Hyperplasia
Compensatory hyperplasia of erythropoietic cells in the bone marrow occurs, leading to an increase in reticulocyte counts in peripheral blood, sometimes reaching levels of 5%–20%. Nucleated RBCs may be observed on peripheral blood smears. In cases of severe hemolysis, immature granulocytes may also be present. Bone marrow smear analysis typically shows hyperactive hematopoiesis with an increased proportion of erythroid cells, predominantly intermediate and late erythroblasts, with a possible inversion of the granulocyte-to-erythrocyte ratio. Occasionally, red blood cells may contain nuclear remnants such as Howell-Jolly bodies and Cabot rings.
Clinical Manifestations
Acute HA, predominantly resulting from intravascular hemolysis, often has a sudden onset, with clinical manifestations including severe lower back and limb pain, headache, vomiting, chills, high fever, pallor, hemoglobinuria, and jaundice. In severe cases, peripheral circulatory failure and acute renal failure may develop.
Chronic HA, typically arising from extravascular hemolysis, manifests as anemia, jaundice, and splenomegaly. Prolonged hyperbilirubinemia may lead to complications such as cholelithiasis and hepatic dysfunction. During the course of chronic hemolysis, triggers such as infections may exacerbate the hemolysis, causing hemolytic or aplastic crises. In cases of chronic severe hemolytic anemia, yellow marrow in long bones may transform into red marrow, accompanied by marrow cavity expansion, cortical bone thinning, and skeletal deformities. Extramedullary hematopoiesis may lead to hepatosplenomegaly.
Laboratory Investigations
In addition to general laboratory tests for anemia, such as routine bloodwork, HA laboratory diagnostics can be categorized into three aspects based on the aforementioned pathogenesis:
- Tests to assess increased RBC destruction.
- Tests to evaluate compensatory erythropoietic hyperplasia.
- Tests to identify the specific cause of hemolysis.
The first two categories are considered screening tests for HA and are used to determine the presence and location of hemolysis. The third category involves specialized tests for HA, which are discussed in specific subsections.
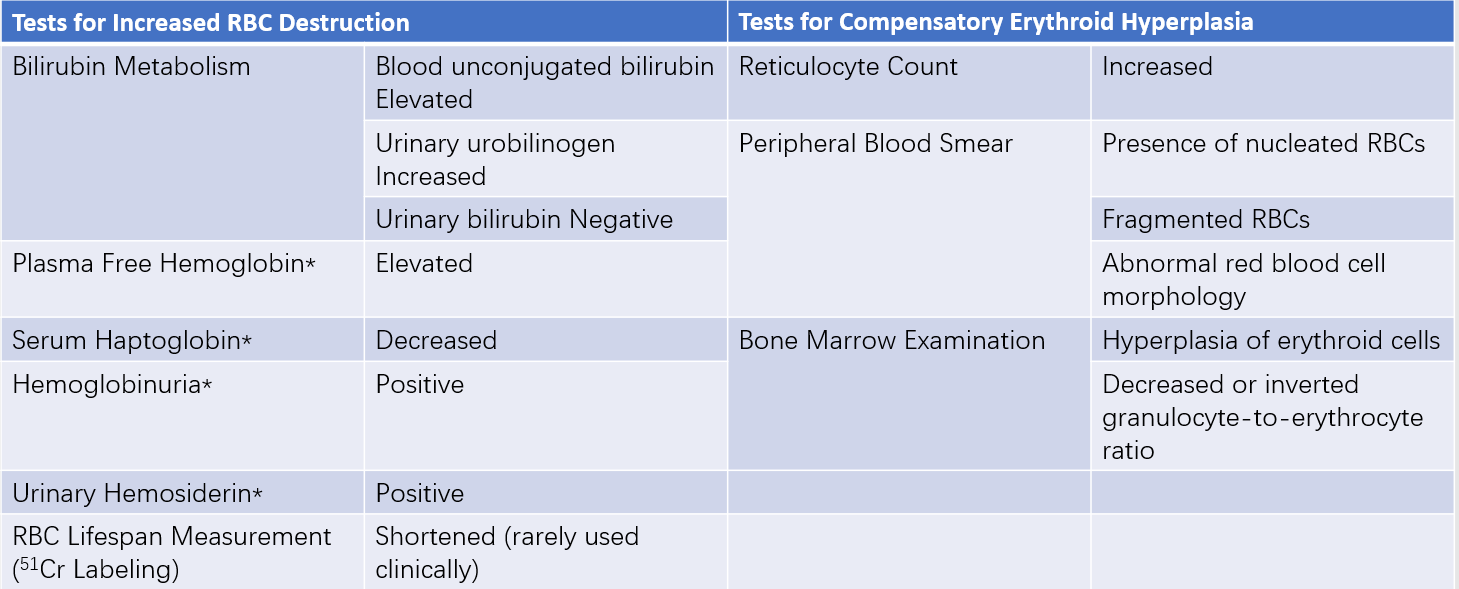
Table 1 Screening tests for hemolytic anemia
Note: Tests marked with * are specific for intravascular hemolysis.
Diagnosis
The diagnosis of hemolytic anemia (HA) can be established based on clinical manifestations, laboratory evidence of anemia, increased RBC destruction, and compensatory hyperplasia of erythroid cells in the bone marrow. The site of hemolysis can also be identified. Detailed medical history and specialized diagnostic tests for HA help ascertain the underlying cause and specific type of HA.
Differential Diagnosis
Several clinical conditions may be mistaken for HA:
Anemia with Increased Reticulocyte Counts
Examples include the early recovery stages of blood loss anemia, iron deficiency anemia, or megaloblastic anemia.
Non-Bilirubinuric Jaundice
Examples include familial non-hemolytic jaundice such as Gilbert syndrome.
Anemia with Nucleated Red and Granulocytic Precursors and Mild Reticulocyte Elevation
Examples include conditions such as bone marrow metastases.
Although these situations may resemble HA, they are not caused by hemolysis and lack laboratory evidence of increased RBC destruction, which facilitates differentiation.
Treatment
Etiological Treatment
Treatment targeting the underlying mechanism of HA, such as glucocorticoids, splenectomy, or C5 inhibitors, is addressed in specific subsections.
Symptomatic Treatment
Management of anemia and complications related to HA includes red blood cell transfusion, correction of acute renal failure, shock, and electrolyte imbalances, as well as measures to prevent thrombus formation.