- Edited
Acute leukemia (AL) is a malignant clonal disorder originating from hematopoietic stem/progenitor cells. During its onset, abnormal blasts and immature cells (leukemia cells) proliferate extensively in the bone marrow, suppressing normal hematopoiesis and infiltrating various organs, such as the liver, spleen, and lymph nodes. Clinical symptoms include anemia, bleeding, infection, and organ infiltration.
Classification
The classification of AL includes the French-American-British (FAB) system and the World Health Organization (WHO) system, with WHO classification being the most commonly used in clinical practice. The FAB classification is based on the observation and quantification of bone marrow cell morphology and cytochemical staining, serving as a fundamental diagnostic criterion. In contrast, the WHO classification integrates morphology, immunology, cytogenetics, and molecular biology (collectively known as MICM) features, providing guidance for treatment planning and prognosis assessment.
FAB Classification of AL
FAB Classification of AML
M0 (Minimally Differentiated Acute Myeloblastic Leukemia):
Bone marrow blasts >30%; no azurophilic granules or Auer rods; prominent nucleoli. Less than 3% of cells are positive for myeloperoxidase (MPO) or Sudan Black B under a light microscope. Electron microscopy shows MPO positivity. Myeloid antigens such as CD33 or CD13 may be positive, while lymphoid and platelet antigens are typically negative.
M1 (AML Without Maturation):
Myeloblasts (Type I + Type II; Type I lacks granules while Type II has a few granules) account for more than 90% of non-erythroid nucleated cells (NEC) in the bone marrow. At least 3% of the cells are MPO-positive.
M2 (AML with Maturation):
Myeloblasts constitute 30%–89% of NEC in the bone marrow, while other granulocytes account for at least 10%. Monocytes make up less than 20%.
M3 (Acute Promyelocytic Leukemia, APL):
The bone marrow is primarily composed of hypergranular promyelocytes, which constitute at least 30% of NEC.
M4 (Acute Myelomonocytic Leukemia, AMMoL):
Blasts account for more than 30% of NEC in the bone marrow. Granulocytes at all maturation stages constitute at least 20%, and monocytes at all stages of maturation also constitute at least 20%.
M4Eo (AML with Eosinophilia):
Characteristics are similar to M4, with eosinophils constituting at least 5% of NEC.
M5 (Acute Monocytic Leukemia, AMoL):
Blasts and promonocytes comprise at least 30% of NEC in the bone marrow, while the sum of blasts, promonocytes, and monocytes accounts for at least 80%. If promonocytes exceed 80%, the subtype is classified as M5a; if less than 80%, it is classified as M5b.
M6 (Erythroleukemia, EL):
Erythroblasts constitute at least 50% of bone marrow cells, with blasts (Type I + Type II) making up at least 30% of NEC.
M7 (Acute Megakaryoblastic Leukemia, AMeL):
Megakaryoblasts account for at least 30% of the bone marrow. Platelet antigens are positive, and platelet peroxidase is also positive.
FAB Classification of ALL
L1:
Blasts and immature lymphocytes are predominantly small cells (diameter ≤12 μm).
L2:
Blasts and immature lymphocytes are predominantly large cells (diameter >12 μm).
L3 (Burkitt Type):
Blasts and immature lymphocytes are uniform in size, predominantly large cells. Cytoplasmic vacuoles are prominent, cytoplasm is deeply basophilic, and staining is intense.
WHO Classification of AL
WHO Classification of AML (Fifth Edition)
AML with Recurrent Genetic Abnormalities:
APL with PML::RARA
AML with RUNX1::RUNX1T1
AML with CBFB::MYH11
AML with DEK::NUP214
AML with RBM15::MRTFA
AML with BCR::ABL1
AML with KMT2A Rearrangement
AML with MECOM Rearrangement
AML with NUP98 Rearrangement
AML with NPM1 Mutation
AML with CEBPA Mutation
AML, Myelodysplasia-Related
AML with Other Specific Genetic Alterations
AML Defined by Differentiation:
Minimally Differentiated AML
AML Without Maturation
AML With Maturation
Acute Basophilic Leukemia
Acute Myelomonocytic Leukemia
Acute Monocytic Leukemia
Acute Erythroid Leukemia
Acute Megakaryoblastic Leukemia
WHO Classification of ALL (Fifth Edition)
Precursor B-Lymphoblastic Leukemia:
B-ALL, Not Otherwise Specified (NOS)
B-ALL with High-Hyperdiploidy
B-ALL with Hypodiploidy
B-ALL with Intrachromosomal Amplification of Chromosome 21 (iAMP21)
B-ALL with BCR::ABL1 Fusion
B-ALL with BCR::ABL1-like Features
B-ALL with KMT2A Rearrangement
B-ALL with ETV6::RUNX1 Fusion
B-ALL with ETV6::RUNX1-like Features
B-ALL with TCF3::PBX1 Fusion
B-ALL with IGH::IL3 Fusion
B-ALL with TCF3::HLF Fusion
B-ALL with Other Specific Genetic Abnormalities
Precursor T-Lymphoblastic Leukemia:
T-ALL, Not Otherwise Specified (NOS)
Early T-Precursor Lymphoblastic Leukemia (ETP-ALL)
Clinical Manifestations
The onset of acute leukemia (AL) varies in intensity, ranging from sudden, acute presentations to more gradual developments. Patients with an acute onset might display high fever or severe bleeding, whereas those with a slower onset often present with pale complexion, skin purpura, excessive menstrual bleeding, or uncontrollable bleeding after tooth extraction, leading to their diagnosis during medical consultation.
Manifestations of Suppressed Normal Bone Marrow Hematopoietic Function
Anemia
Some patients, particularly those with a short disease course, may not exhibit anemia. However, more than half of patients present with severe anemia at diagnosis, particularly those secondary to myelodysplastic syndromes (MDS).
Fever
Fever serves as an early symptom in approximately half of patients. The fever may be mild or reach high temperatures exceeding 39–40°C, often accompanied by chills and sweating. While leukemia itself can cause fever, concurrent infections must also be considered. Infections can involve various sites, with stomatitis, gingivitis, and pharyngitis being most common. These may progress to ulcers or necrosis. Lung infections, perianal inflammation, and pararectal abscesses are also frequent and can lead to bloodstream infections in severe cases. The most common pathogens are gram-negative bacilli, such as Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, and Acinetobacter species. Gram-positive cocci, such as Staphylococcus aureus, coagulase-negative staphylococci, and enterococci, are being observed more frequently. Long-term antibiotic use and neutropenia may predispose patients to fungal infections, including Candida, Aspergillus, and Cryptococcus species. Viral infections, such as herpes simplex virus, varicella-zoster virus, and cytomegalovirus, are also possible due to immune deficiency, with Pneumocystis jirovecii pneumonia being a rare occurrence.
Bleeding
Bleeding is an early symptom in about 40% of cases. It can occur throughout the body, manifesting as petechiae and ecchymoses on the skin, epistaxis, gingival bleeding, or menorrhagia. Retinal hemorrhages may cause visual disturbances. Acute promyelocytic leukemia (APL) is prone to coagulation abnormalities, leading to widespread systemic bleeding. Intracranial hemorrhage may result in headache, vomiting, anisocoria, and even coma or death. Data indicate that 62.24% of deaths in AL are attributed to bleeding, with 87% of these from intracranial hemorrhage. Major causes of bleeding include vascular infiltration by large numbers of leukemic cells, thrombocytopenia, coagulation abnormalities, and infection.
Manifestations of Leukemic Cell Proliferation and Infiltration
Lymphadenopathy and Hepatosplenomegaly
Lymph node enlargement is more commonly observed in acute lymphoblastic leukemia (ALL), with mediastinal lymphadenopathy frequently associated with T-ALL. Hepatosplenomegaly is generally mild to moderate, and massive splenomegaly is rare, except in cases of CML in blast crisis.
Bones and Joints
Localized tenderness over the lower sternum is commonly reported. Joint pain and bone pain are frequent, particularly among children. Bone marrow necrosis may cause severe bone pain.
Ocular Involvement
Some cases of acute myeloid leukemia (AML) may present with granulocytic sarcoma (also known as chloroma), which frequently involves the periosteum, especially around the orbit, causing proptosis, diplopia, or vision loss.
Oral and Skin Involvement
Leukemic cell infiltration, especially in M4 and M5 subtypes, may lead to gingival hyperplasia and swelling. Leukemia cutis can manifest as bluish-gray papular lesions, localized skin thickening, or purple-blue nodules.
Central Nervous System (CNS)
The CNS is the most common extramedullary infiltration site in leukemia. Due to the inability of most chemotherapeutic drugs to effectively cross the blood-brain barrier, residual leukemic cells in the CNS can lead to central nervous system leukemia (CNSL). Symptoms may range from mild manifestations like headache and dizziness to severe ones like vomiting, neck stiffness, seizures, and coma. CNSL can occur at any stage of the disease and is most commonly seen during remission following treatment, particularly in ALL, with children most frequently affected. Other subtypes such as M4, M5, and M2 are also occasionally involved.
Testicular Involvement
The testicles, particularly unilaterally, may show painless enlargement, although the contralateral testis may also reveal leukemic cell infiltration on biopsy despite appearing unaffected. Testicular leukemia is most commonly observed in pediatric and young adult ALL patients during chemotherapy remission and represents the second most common site of extramedullary relapse after the CNS.
In addition, leukemia can infiltrate other tissues and organs. The lungs, heart, gastrointestinal tract, and genitourinary systems may all be affected.
Laboratory Examinations
Peripheral Blood
Most patients with acute leukemia (AL) show elevated white blood cell (WBC) counts; those with WBC >10×109/L are classified as having hyperleukocytic leukemia. However, some individuals may have normal or reduced WBC counts, with levels as low as <1.0×109/L, termed aleukemic leukemia. Peripheral blood smears often demonstrate varying degrees of blasts and immature cells, although blasts may not be detectable in certain cases of aleukemic leukemia. Anemia of varying severity is frequently observed, and a minority of patients exhibit anisocytosis or have nucleated red blood cells identified in blood smears. Around 50% of patients have platelet counts below 60×109/L, with severe thrombocytopenia typically observed in advanced stages.
Bone Marrow
Bone marrow examination is the main diagnostic tool for AL. According to the FAB classification, the diagnosis of AL is established when blasts comprise ≥30% of nucleated cells in the bone marrow (ANC). The WHO classification has lowered this threshold to ≥20% and has removed the ≥20% blast threshold for AML with recurrent genetic abnormalities (except for AML with BCR::ABL1 fusion and AML with CEBPA mutations).
In most cases, significant hypercellularity is observed in the bone marrow, with a predominance of blasts. However, in some cases, marrow hypoplasia occurs, which is referred to as hypoplastic AL.
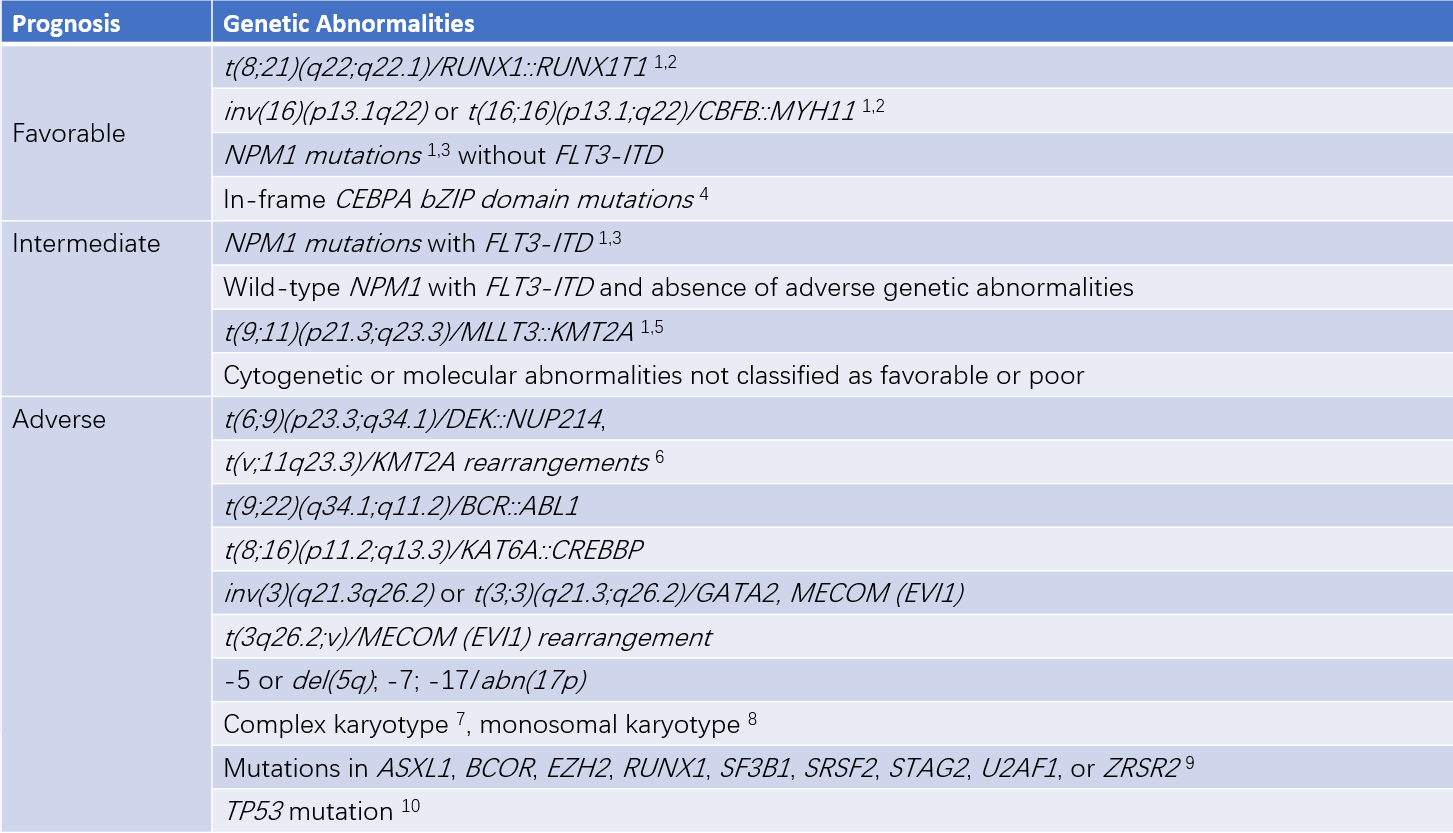
Cytochemistry
Cytochemical staining is applied to assist in the morphological differentiation of various types of leukemia.
Table 1 Cytochemical differentiation of common acute leukemias (AL)
Immunological Tests
The immunophenotype of leukemic cells is determined based on the expression of lineage-associated antigens. Hematopoietic stem/progenitor cells express CD34, while acute promyelocytic leukemia (APL) cells frequently express CD13, CD33, and CD117 but do not express HLA-DR or CD34. APL cells may also express CD9.
Acute mixed-lineage leukemia includes acute biphenotypic leukemia (leukemic cells co-express both myeloid and lymphoid antigens) and biclonal leukemia (two distinct leukemic cell clones derived from separate progenitor cells, each expressing either myeloid or lymphoid antigens). A score greater than 2 for both myeloid and one lymphoid lineage is indicative of these leukemias.
Table 2 European group for the immunological classification of leukemias (EGIL, 1998)
Note:
"Cy" refers to markers detected intracellularly (cytoplasmic),
"TCR" refers to T-cell receptor.
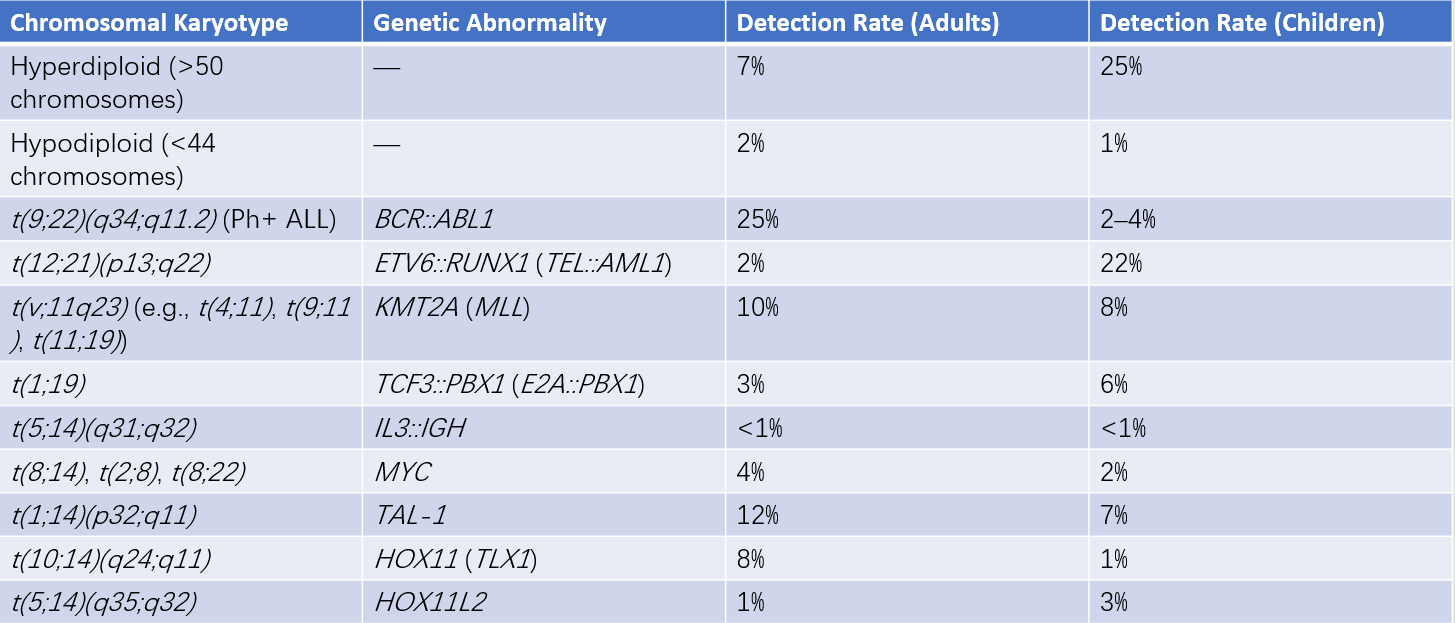
Cytogenetics and Molecular Biology
Leukemia is often associated with specific cytogenetic (chromosomal karyotype) and molecular genetic abnormalities (e.g., fusion genes, gene mutations). For instance, approximately 99% of APL cases involve the t(15;17)(q22;q12) translocation, which results in the formation of the PML::RARA fusion gene from the PML gene on chromosome 15 and the RARA gene on chromosome 17. This genetic rearrangement underpins the pathogenesis of APL and the effectiveness of treatment with all-trans retinoic acid and arsenic-based therapies.
Table 3 Prognostic significance of common genetic abnormalities in AML
Note:
1, The results are primarily based on observations in patients receiving intensive therapy. Initial risk categories may change during treatment according to analyses of measurable residual disease.
2, KIT or FLT3 mutations do not alter the risk categories.
3, AML with NPM1 mutations and high-risk cytogenetic abnormalities is classified as an adverse prognosis group.
4, In-frame mutations exclusively affecting the bZIP domain of the CEBPA gene, whether monoallelic or biallelic, are associated with favorable outcomes.
5, The presence of t(9;11)(p21.3;q23.3) takes precedence over rare coexisting adverse prognostic mutations.
6, The analysis excludes partial tandem duplications (PTD) of KMT2A.
7, A complex karyotype is defined as having ≥3 unrelated chromosomal abnormalities without other recurrent genetic abnormalities defining a specific subtype. This excludes hyperdiploid karyotypes with ≥3 trisomies (or polysomies) without structural abnormalities.
8, A monosomal karyotype includes the presence of ≥2 different autosomal monosomies (excluding loss of X or Y chromosomes) or one autosomal monosomy combined with at least one structural chromosomal abnormality, excluding core-binding factor AML.
9, These markers, when co-occurring with favorable prognostic AML subtypes, are not currently considered adverse prognostic markers.
10, Regardless of the TP53 allelic status (whether monoallelic or biallelic mutations), the allelic fraction of TP53 mutations must be at least 10%. TP53 mutations are significantly associated with complex karyotypes and monosomal karyotypes in AML.
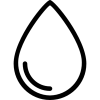
Table 4 Detection rates of common chromosomal and molecular abnormalities in ALL
Note: ALL with t(9;22)(q34;q11.2) is also referred to as Philadelphia chromosome-positive ALL (Ph+ ALL).
Biochemical Tests
Elevated serum uric acid concentrations, particularly during chemotherapy, are common, accompanied by increased uric acid excretion and, in some cases, uric acid crystal formation.
Coagulation abnormalities may be detected in patients with disseminated intravascular coagulation (DIC). Elevated serum lactate dehydrogenase (LDH) levels may also be present.
In cases of central nervous system leukemia (CNSL), cerebrospinal fluid (CSF) reveals increased pressure, elevated WBC counts, elevated protein concentrations, and decreased glucose levels. Leukemic cells may also appear in CSF smears.
Diagnosis and Differential Diagnosis
A leukemia diagnosis is typically straightforward when based on clinical presentation, peripheral blood findings, and bone marrow characteristics. However, due to variability in the MICM (morphology, immunophenotype, cytogenetics, and molecular biology) features of leukemic cells, treatment regimens and prognoses also differ. Comprehensive MICM data should be obtained at the time of initial diagnosis to allow prognosis assessment and treatment guidance. Additionally, careful assessment is required to exclude the following conditions:
Myelodysplastic Neoplasms (MDS)
MDS may be confused with leukemia as its features include dysplastic hematopoiesis, the presence of blasts and immature cells in peripheral blood, pancytopenia, and chromosomal abnormalities. However, the percentage of blasts in the bone marrow is less than 20%.
Leukocytosis Caused by Certain Infections
Infectious mononucleosis can be associated with atypical lymphocytes in the peripheral blood, which differ morphologically from blasts. These cases are characterized by a gradually rising heterophile antibody titer, a short disease course, and spontaneous recovery. Pertussis, infectious lymphocytosis, rubella, and other viral infections may present with increased lymphocytes in peripheral blood, although the lymphocyte morphology remains normal and the disease course is generally benign. Increased blasts and immature cells in the bone marrow are not observed in these conditions.
Megaloblastic Anemia
Megaloblastic anemia can occasionally resemble erythroleukemia. However, in megaloblastic anemia, there is no increase in blasts in the bone marrow, and immature red blood cells usually show a negative periodic acid-Schiff (PAS) reaction. Treatment with folic acid or vitamin B12 is effective.
Recovery from Agranulocytosis
During recovery from drug- or infection-induced agranulocytosis, increased promyelocytes and myelocytes may appear in the bone marrow. However, this condition often has an identifiable cause, normal platelet counts, absence of Auer rods in the blasts, and no chromosomal abnormalities. Normal maturation and recovery of bone marrow granulocytes occur within a short period.
Treatment
Treatment plans are developed based on the patient's MICM (morphology, immunophenotype, cytogenetics, and molecular biology) characteristics and clinical presentation, as well as the patient’s preferences and financial capacity. Prognostic risk stratification is performed to guide the selection and design of a comprehensive and systematic treatment regimen. To address the need for frequent interventions and to reduce patient discomfort from repeated needle punctures, the insertion of a central venous catheter is typically recommended. Patients eligible for allogeneic hematopoietic stem cell transplantation (allo-HSCT) require blood tests for HLA typing.
General Treatment
Urgent Management of Hyperleukocytosis
White blood cell (WBC) counts exceeding 100×109/L in the circulating blood can lead to leukostasis syndrome, with clinical manifestations such as respiratory distress, hypoxemia, confusion, slurred speech, and intracranial hemorrhage. Pathological findings often demonstrate simultaneous leukemic thrombosis and bleeding. High WBC counts increase both early mortality and the risk of extramedullary leukemia and relapse. For WBC >100×109/L, leukapheresis can be performed to rapidly reduce WBC counts (not typically recommended for acute promyelocytic leukemia, APL). This is accompanied by alkalization, hydration, and chemotherapy. Short pre-chemotherapy regimens may also be used: ALL patients may receive dexamethasone at 10 mg/m2 intravenously, and AML patients may be given hydroxyurea at 1.5–2.5 g every 6 hours (total dose 6–10 g/day) for approximately 36 hours before initiating combination chemotherapy. Complications such as hyperuricemia, acidosis, electrolyte imbalance, and coagulation abnormalities induced by leukemic cell lysis should be closely monitored and prevented.
Infection Prevention and Management
Leukemia patients, particularly those undergoing chemotherapy or radiotherapy, often experience prolonged periods of neutropenia. During this time, patients benefit from staying in laminar airflow isolation rooms or disinfected isolation wards. Granulocyte colony-stimulating factor (G-CSF) reduces the duration of neutropenia and is recommended for ALL patients, as well as for elderly AML patients, those undergoing intensive chemotherapy, or those with infections. For febrile episodes, bacterial cultures and antimicrobial susceptibility testing are essential, and empirical antibiotic therapy is initiated promptly.
Transfusion Support
Severe anemia may require oxygen supplementation and transfusion of packed red blood cells, aiming to maintain hemoglobin levels above 80 g/L. However, red blood cell transfusion is typically avoided during leukostasis to prevent further increases in blood viscosity. Severe thrombocytopenia requires transfusion of single-donor platelet concentrates. To reduce the occurrence of ineffective transfusions and febrile reactions caused by alloimmune responses, leukocyte-filtered blood components are generally used. To prevent transfusion-associated graft-versus-host disease (TA-GVHD), irradiation (25–30 Gy) of cellular blood products is necessary to inactivate lymphocytes prior to transfusion.
Prevention and Management of Hyperuricemic Nephropathy
The destruction of large numbers of leukemic cells, exacerbated during chemotherapy, causes serum and urinary uric acid levels to rise, potentially leading to uric acid crystal deposition in renal tubules. Hyperuricemic nephropathy may result. Patients are encouraged to maintain high fluid intake, preferably with continuous intravenous hydration over 24 hours, to achieve a urine output of over 100 ml/m2 per hour and to keep the urine alkaline. Concurrent administration of allopurinol at 100 mg, three times a day, can inhibit uric acid synthesis. Severe skin allergic reactions may occur with allopurinol use in rare cases and should be monitored. Acute renal failure management protocols are required for patients experiencing oliguria, anuria, or renal dysfunction.
Nutritional Support
Leukemia is a highly consumptive disease, particularly during chemotherapy or radiotherapy, which can lead to mucositis and gastrointestinal dysfunction. Adequate nutritional supplementation and maintenance of water and electrolyte balance are crucial. Patients benefit from high-protein, high-calorie, and easily digestible diets. In some cases, parenteral nutritional support is necessary.
Anti-Leukemia Therapy
The first phase of anti-leukemia treatment focuses on induction therapy, primarily involving combination chemotherapy. The goal is to rapidly achieve complete remission (CR), which is defined by the disappearance of symptoms and signs of leukemia, the absence of blasts in peripheral blood or extramedullary sites, the recovery of trilineage hematopoiesis in the bone marrow (with blasts <5%), absolute neutrophil counts exceeding 1.0×109/L, and platelet counts ≥100×109/L.
Once CR is achieved, treatment enters the second phase, known as post-remission therapy. The primary modalities for this phase are chemotherapy and hematopoietic stem cell transplantation (HSCT). After induction therapy achieves CR, the leukemic cell burden decreases from an initial level of 1010–1012/L to approximately 108–109/L. These remaining leukemic cells are referred to as measurable residual disease (MRD). MRD levels are predictive of relapse and must be regularly monitored. Sustained MRD negativity offers the possibility of long-term disease-free survival (DFS) and even cure (DFS of more than 10 years).
Treatment of ALL
Advances in chemotherapy regimens have significantly improved outcomes for pediatric ALL, with long-term DFS rates now exceeding 80%. Adolescent ALL patients generally benefit from treatment with pediatric protocols. In adults, with enhanced supportive care, multi-drug combination therapies, high-dose chemotherapy regimens, and the application of HSCT, CR rates have reached 80–90%, with considerable improvement in prognosis. Treatment options for ALL require consideration of multiple factors, including patient age, ALL subtype, MRD status after therapy, the availability of stem cell donors, and targeted therapeutic agents.
Induction Therapy for Remission
The VP regimen, which consists of vincristine (VCR) and prednisone (P), is the basic treatment protocol for acute lymphoblastic leukemia (ALL). The VP regimen achieves a complete remission (CR) rate of approximately 50% in adult ALL, with a CR duration of 3–8 months. Peripheral neuropathy and constipation are the primary toxic side effects of VCR. Adding an anthracycline (e.g., daunorubicin [DNR]) to the VP regimen creates the DVP protocol, increasing the CR rate to over 70%, although anthracycline-associated cardiotoxicity requires caution. The DVLP regimen, which incorporates L-asparaginase (L-Asp) or pegylated asparaginase (PEG-Asp) into the DVP regimen, is currently a commonly used induction protocol for ALL. L-Asp and PEG-Asp improve disease-free survival (DFS) rates but are associated with side effects such as liver dysfunction, pancreatitis, reduced synthesis of clotting factors and albumin, and hypersensitivity reactions.
Adding additional agents to the DVLP regimen, such as cyclophosphamide (CTX) or cytarabine (Ara-C), or employing the Hyper-CVAD protocol (consisting of alternating cycles of CTX, VCR, doxorubicin, and dexamethasone in the first phase, followed by high-dose methotrexate [HD-MTX] and Ara-C in the second phase), can further improve CR rates and DFS in some ALL cases.
Post-Remission Treatment
Post-remission therapy generally consists of two phases: intensive consolidation and maintenance therapy. Intensive consolidation therapy primarily involves chemotherapy or hematopoietic stem cell transplantation (HSCT). Chemotherapy often includes cyclic repetition of induction regimens, supplemented periodically with other intensified protocols. Higher dosages of chemotherapeutic agents are commonly used, with alternating and rotating agents to minimize cumulative toxicity. High-dose methotrexate (HD-MTX), Ara-C, 6-mercaptopurine (6-MP), and L-Asp or PEG-Asp are frequently employed during this phase. HD-MTX is associated with mucositis and liver or kidney dysfunction, requiring adequate hydration, alkalization, and timely rescue therapy with leucovorin during treatment.
For ALL (excluding mature B-ALL), even with intensive induction and consolidation therapy, maintenance therapy is often necessary. Maintenance therapy commonly includes oral 6-MP and MTX combined with intermittent VP regimen chemotherapy, which has proven effective. In the absence of allo-HSCT, consolidation and maintenance therapy for ALL generally last 2–3 years, with regular monitoring of measurable residual disease (MRD). The intensity and duration of consolidation and maintenance therapy are determined based on ALL subtypes. For MRD-positive B-ALL patients across all age groups, therapies such as CD19/CD3 bispecific antibodies to eliminate residual leukemic cells followed by allo-HSCT may be considered.
For Philadelphia chromosome-positive (Ph+) ALL, combining tyrosine kinase inhibitors (TKIs, e.g., imatinib or dasatinib) with induction chemotherapy improves CR rates to 90–95%. Continuous TKI use is recommended until the completion of maintenance therapy. Incorporating allo-HSCT with TKI therapy further enhances survival outcomes and quality of life.
Prevention and Treatment of Central Nervous System Leukemia (CNSL) and Testicular Leukemia
Due to the blood-brain barrier and blood-testis barrier, many chemotherapeutic agents cannot adequately penetrate these "sanctuaries" where leukemic cells reside. Effective management of sanctuary sites is an indispensable component of acute leukemia (AL) therapy, especially for ALL. CNSL prevention is typically integrated throughout the entire course of ALL treatment and includes strategies such as craniospinal irradiation, intrathecal chemotherapy (e.g., MTX, Ara-C, glucocorticoids), and/or high-dose systemic chemotherapy (e.g., HD-MTX, Ara-C). Craniospinal irradiation demonstrates efficacy but is often limited by adverse effects such as cognitive impairment, secondary malignancies, endocrine disorders, and neurotoxicity (e.g., leukoencephalopathy). Early intensive systemic therapy combined with intrathecal chemotherapy is currently favored for CNSL prevention, with craniospinal irradiation reserved for salvage treatment when CNSL occurs. In cases of testicular leukemia, bilateral irradiation and systemic chemotherapy are necessary, even when disease involvement is unilateral.
Relapse is defined as the reappearance of leukemic cells in the peripheral blood or a blast percentage exceeding 5% in the bone marrow after CR (excluding other causes such as bone marrow recovery following consolidation therapy). Leukemic infiltration at extramedullary sites can also signal relapse, which most commonly occurs within two years after CR, with bone marrow relapse being the most frequent. Salvage therapy options include the original induction chemotherapy regimen, combination regimens containing HD Ara-C, or therapies based on CD19/CD3 bispecific antibodies and CD22 antibody-drug conjugates. However, once ALL relapses, the duration of the second remission is typically short, and long-term survival rates are low. Chimeric antigen receptor T-cell (CAR-T) therapies targeting CD19 allow approximately 90% of CD19-positive relapsed ALL patients to achieve CR. Currently, CAR-T cells targeting CD19, CD22, or CD20 (either with single or dual targets) are frequently used to treat B-ALL, while CAR-T therapies targeting CD7 for T-ALL remain in clinical trials.
Extramedullary relapse commonly manifests as CNSL, but isolated extramedullary relapse often coincides with detectable MRD in the bone marrow, and subsequent hematologic relapse typically follows. Systemic chemotherapy is therefore essential in addition to localized therapy for extramedullary relapse.
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is critical for curing adult ALL, enabling long-term survival in 40–65% of patients. Indications for allo-HSCT include:
- Relapsed or refractory ALL.
- ALL in second CR (CR2).
- High-risk ALL in first CR (CR1), such as patients with cytogenetic abnormalities (e.g., Ph+ chromosome, hypodiploidy), those with MLL gene rearrangements, WBC ≥30×109/L in pre-B ALL, WBC ≥100×109/L in T-ALL, time to achieve CR exceeding 4–6 weeks, or those with persistent or increasing MRD during consolidation and maintenance therapy.
Treatment of Acute Myeloid Leukemia (AML)
In recent years, significant advances in the prognosis of AML patients under 60 years of age have been achieved due to intensive chemotherapy, hematopoietic stem cell transplantation (HSCT), and improved supportive care. Approximately 30–50% of AML (non-APL) patients have the potential for long-term survival.
Induction Therapy to Achieve Remission
AML (non-APL):
The standard chemotherapy regimen consists of an anthracycline combined with standard-dose cytarabine (Ara-C), commonly referred to as the 3+7 protocol. The most frequently used protocols include IA (idarubicin, IDA) and DA (daunorubicin, DNR), with overall complete remission (CR) rates of 50–80% for patients under 60 years of age. Under effective supportive care, both IDA at 12 mg/(m2·day) in the IA protocol and DNR at 60–90 mg/(m2·day) in the DA protocol demonstrate high CR rates.
Studies pioneered the substitution of IDA or DNR with homoharringtonine (HHT) to form the HA protocol for AML induction therapy, with CR rates of 60–65%. HA combined with anthracyclines such as DNR or aclarubicin (Acla), forming protocols like HAD and HAA, can further improve CR rates.
For elderly patients or those ineligible for intensive chemotherapy, low-intensity regimens, such as hypomethylating agents combined with BCL-2 inhibitors, achieve remission induction rates comparable to the standard 3+7 protocol. Targeted agents such as FLT3 inhibitors and IDH inhibitors, when used in patients carrying these specific mutations, improve remission rates. For younger patients, intermediate- or high-dose Ara-C combined with anthracyclines can extend disease-free survival but does not necessarily increase the CR rate. Patients who achieve CR after one treatment cycle typically have longer disease-free survival, while those who remain in remission failure after two cycles of standard chemotherapy are likely to have primary resistance, requiring a change in chemotherapy regimen or allo-HSCT.
APL:
Current treatment predominantly uses all-trans retinoic acid (ATRA) combined with arsenic agents, such as arsenic trioxide (ATO), and, if necessary, includes hydroxyurea or anthracyclines to control white blood cell counts. ATRA targets RARA and induces differentiation and maturation of APL cells carrying the PML::RARA fusion gene at doses of 20–45 mg/(m2·day). ATO acts on PML and induces APL cell differentiation at low doses and apoptosis at high doses.
Differentiation syndrome (also known as retinoic acid syndrome) may occur during treatment, particularly in cases with high initial white blood cell counts or rapid increases following therapy. This syndrome, thought to result from massive cytokine release and increased adhesion molecule expression, manifests with fever, musculoskeletal pain, respiratory distress, pulmonary interstitial infiltration, pleural effusion, pericardial effusion, weight gain, hypotension, acute renal failure, and potentially death. At the onset of any of these symptoms, corticosteroid therapy, oxygen supplementation, and diuretics are required, and ATRA treatment may be temporarily discontinued. In addition to differentiation syndrome, other adverse effects of ATRA include headache, intracranial hypertension, and hepatic dysfunction, while ATO is associated with liver dysfunction and QT interval prolongation on electrocardiograms. For APL with coagulopathy and bleeding, proactive transfusion of platelets, fresh frozen plasma, and cryoprecipitates can reduce early mortality due to hemorrhage.
Post-Remission Treatment
The key features of post-remission therapy for AML are as follows:
- The incidence of central nervous system leukemia (CNSL) in AML is less than 3%. For newly diagnosed patients with WBC counts ≥40×10⁹/L, extramedullary disease, M4/M5 subtype, or t(8;21) or inv(16), cerebrospinal fluid testing should be performed after CR, followed by at least one prophylactic intrathecal therapy for CNSL screening. In APL patients, at least three prophylactic intrathecal therapies are recommended after CR.
- Therapy duration for AML (non-APL) is significantly shorter compared to ALL.
- Patients with APL achieving molecular remission can alternate medications such as ATRA and arsenic agents for maintenance therapy over approximately two years, with periodic monitoring to sustain PML::RARA fusion gene negativity.
For AML patients under 60 years old, treatment selection depends on risk stratification:
- Poor-risk group: Allo-HSCT is the first choice.
- Favorable-risk group (non-APL): High-dose Ara-C-based chemotherapy is preferred; relapse may warrant allo-HSCT.
- Intermediate-risk group: Both allo-HSCT and high-dose Ara-C-based chemotherapy are viable options.
For patients ineligible for allo-HSCT within the unfavorable-risk, certain favorable-risk, and intermediate-risk groups, autologous HSCT may be considered. Patients who cannot be stratified should follow treatment protocols for the intermediate-risk group, but those with initial WBC counts ≥100×109/L should be treated as part of the poor-risk group.
In cases where patients are unable to undergo these treatment options due to age, comorbidities, or other factors, conventional chemotherapy regimens with various agents may be rotated for consolidation and maintenance. However, this approach achieves long-term survival in only 10–15% of patients.
High-dose Ara-C is associated with notable complications, including cerebellar ataxia, necessitating discontinuation upon occurrence. Other common side effects include rash, fever, and conjunctivitis, for which corticosteroid prophylaxis is often used.
Treatment of Relapsed and Refractory AML
Treatment options include:
- Targeted agents (e.g., FLT3 inhibitors, IDH inhibitors).
- Combination regimens with intermediate- or high-dose Ara-C.
- Combination chemotherapy with non-cross-resistant novel agents.
- HSCT.
- Enrollment in clinical trials.
Patients achieving CR following re-induction therapy should proceed to allo-HSCT as soon as possible. For relapsed APL, re-induction therapy with ATO ± ATRA ± anthracycline-based chemotherapy can be utilized. Patients achieving CR and attaining fusion gene negativity may undergo autologous HSCT or arsenic-based consolidation therapy (for those ineligible for transplantation). Patients with persistent gene positivity should consider allo-HSCT or clinical trial enrollment.
Treatment of Acute Leukemia (AL) in the Elderly
In most patients over 60 years of age with acute leukemia, chemotherapy requires dose reduction to lower the mortality rate associated with treatment. A small subset of patients with robust physical fitness and good supportive care may undergo treatment regimens similar to those used for younger patients. Patients under 70 years of age with generally good overall condition, preserved function of critical organs, poor prognostic factors, and suitable donors may be eligible for reduced-intensity conditioning (RIC) allogeneic hematopoietic stem cell transplantation (allo-HSCT). Patients with AL secondary to myelodysplastic syndrome (MDS), AL caused by exposure to physical or chemical factors, therapy-resistant AL, organ dysfunction, or AL associated with unfavorable cytogenetics or gene mutations require more emphasis on individualized treatment approaches.
In recent years, the combined use of BCL-2 inhibitors and hypomethylating agents, CD3/CD19 bispecific antibodies, and targeted therapies such as tyrosine kinase inhibitors (TKIs) has been transforming the treatment landscape for elderly patients.
Prognosis
Without specific treatment, the average survival time for AL is approximately three months, with some cases experiencing death just days after diagnosis. However, modern treatments have enabled many patients to achieve long-term survival.
For acute lymphoblastic leukemia (ALL), patients between 1–9 years of age, with white blood cell counts <50×109/L, and associated with hyperdiploidy or t(12;21), have the best prognosis, with long-term disease-free survival or even cure rates exceeding 80%. Acute promyelocytic leukemia (APL) has a favorable prognosis if early mortality can be avoided, and many patients can be cured.
Elderly patients and those with high leukocyte counts in AL have poorer prognoses. Chromosome abnormalities and certain molecular markers provide independent prognostic information. Secondary AL, relapsed AL, AL with multidrug resistance, AL requiring multiple chemotherapy cycles to achieve remission, and AL with extramedullary involvement are associated with worse outcomes.
It should also be noted that the prognostic significance of some indicators changes with advancements in treatment strategies. For example, with the advent of tyrosine kinase inhibitors, the prognosis of Philadelphia chromosome-positive (Ph+) ALL has gradually improved.