Chronic myelogenous leukemia (CML), also known as chronic granulocytic leukemia, is a malignant myeloproliferative tumor originating from multipotent hematopoietic stem cells. It is classified as an acquired malignant clonal hematopoietic stem cell disorder primarily involving the myeloid lineage. Peripheral blood granulocytes are markedly increased, and the presence of the Philadelphia chromosome (Ph) and/or the BCR::ABL1 fusion gene can be detected in the affected cell lineages. The disease follows a relatively slow progression and is frequently characterized by splenomegaly. The natural history of CML is divided into three phases: the chronic phase (CP), the accelerated phase (AP), and the blastic phase or blast crisis (BP/BC).
Clinical Features and Laboratory Findings
The onset is insidious, and early stages often lack noticeable symptoms. Patients may be diagnosed during routine health checkups or during evaluations for unrelated conditions when abnormalities in blood counts or splenomegaly are discovered.
Chronic Phase (CP)
The chronic phase usually lasts 1–4 years. Symptoms related to metabolic hyperactivity, such as fatigue, low-grade fever, diaphoresis, and weight loss, are commonly observed. Splenomegaly may result in a subjective feeling of heaviness or fullness in the left upper abdomen. Splenomegaly is often the most prominent feature, with the spleen frequently extending to or beyond the umbilicus, being firm, smooth, and non-tender. Splenic infarctions, if present, may cause tenderness in the splenic area along with a friction rub. Hepatomegaly occurs less frequently. Some patients present with tenderness in the mid-lower sternum. Severe leukocytosis may lead to fundus congestion or retinal hemorrhages, and extreme leukocytosis may result in leukostasis syndrome.
Peripheral Blood Counts
Leukocyte counts are markedly elevated, commonly exceeding 20×109/L and sometimes surpassing 100×109/L. Peripheral smears show a significant increase in granulocytes at various stages of maturation, predominantly myelocytes, metamyelocytes, and band cells. Blast cells (types I+II) are less than 10%. Eosinophils and basophils are elevated, particularly the latter, which aids in diagnosis. Platelets may remain at normal levels, increase in about half of cases, and progressively decrease in later stages, often accompanied by anemia.
Neutrophilic Alkaline Phosphatase (NAP)
Activity is reduced or completely negative. Effective treatment restores NAP activity, while relapses result in its decline. Bacterial infections may cause a mild elevation.
Bone Marrow Findings
Hypercellularity is observed, with marked granulocytic hyperplasia, leading to a prominently increased myeloid-to-erythroid ratio. There is an increased presence of myelocytes, metamyelocytes, and band cells, while blast cells remain less than 10%. Eosinophils and basophils are elevated. Erythropoiesis appears relatively decreased. Megakaryocytes may remain normal or increase, while they decline in later stages. Occasionally, Gaucher cells are observed.
Cytogenetic and Molecular Testing
Over 95% of CML cases show the presence of the Philadelphia chromosome (a small chromosome 22), identified cytogenetically as t(9;22)(q34;q11). This translocation relocates the ABL1 proto-oncogene from the long arm of chromosome 9 to the breakpoint cluster region (BCR) of the long arm of chromosome 22, forming the BCR::ABL1 fusion gene. The BCR::ABL1 fusion protein, commonly p210, exhibits tyrosine kinase activity. The Ph chromosome is present in granulocytic, erythroid, monocytic, megakaryocytic, and lymphocytic cells. Approximately <5% of cases display BCR::ABL1 positivity without the presence of the Ph chromosome.
Biochemical Findings
Elevated serum and urinary uric acid levels, with increased serum lactate dehydrogenase (LDH), can be seen.
Accelerated Phase (AP)
The duration of the AP can range from a few months to several years. Symptoms may include fever, weakness, progressive weight loss, and bone pain. Anemia and bleeding tendencies gradually emerge, along with persistent or progressive splenomegaly. Medications that were previously effective, such as tyrosine kinase inhibitors (TKIs), lose efficacy. This phase is characterized by ≥10% blasts in the peripheral blood or bone marrow, >20% basophils in the peripheral blood, unexplained progressive thrombocytopenia or thrombocytosis, or secondary chromosomal abnormalities in Ph-positive cells (e.g., +8, double Ph chromosomes, or isochromosome 17q [i(17q)]).
Blastic Phase (BC)
The blastic phase represents the terminal stage of CML and typically resembles acute leukemia. Most cases evolve into acute myeloid leukemia (AML), while a minority progress to acute lymphoblastic leukemia (ALL) or acute monocytic leukemia; rare cases of acute transformations involving megakaryocytic or erythroblastic lineages may occur. The prognosis is extremely poor, with death often occurring within a few months. This phase is defined by >20% blasts in the peripheral blood or bone marrow or extramedullary infiltrates of blast cells.
Diagnosis and Differential Diagnosis
A diagnosis of chronic myelogenous leukemia (CML) can be established when persistent unexplained leukocytosis is accompanied by characteristic changes in peripheral blood counts, bone marrow findings, splenomegaly, and positivity for the Philadelphia chromosome (Ph) and/or the BCR::ABL1 fusion gene. The Ph chromosome can also be detected in 1% of acute myeloid leukemia (AML) cases, 5% of pediatric acute lymphoblastic leukemia (ALL) cases, and 25% of adult ALL cases, necessitating differentiation. Diseases with clinical features similar to CML but lacking the Ph chromosome and BCR::ABL1 fusion gene are categorized as myelodysplastic/myeloproliferative neoplasms. Other conditions requiring differentiation include the following:
Splenomegaly of Other Causes
Conditions such as schistosomiasis, chronic malaria, visceral leishmaniasis, liver cirrhosis, and hypersplenism can all result in splenomegaly but are associated with specific clinical features of their primary diseases. Peripheral blood counts and bone marrow findings lack the characteristic changes of CML, and both the Ph chromosome and BCR::ABL1 fusion gene are absent.
Leukemoid Reactions
Leukemoid reactions typically occur secondary to severe infections or underlying malignant tumors and are accompanied by clinical manifestations of the primary condition. Toxic granules and cytoplasmic vacuoles are often present in granulocytes. Eosinophilia and basophilia are generally absent. Neutrophilic alkaline phosphatase (NAP) reaction is strongly positive. The Ph chromosome and BCR::ABL1 fusion gene are absent, and platelet and hemoglobin levels are usually normal. White blood cell counts normalize following the resolution of the underlying condition.
Myelofibrosis
Primary myelofibrosis is characterized by significant splenomegaly and leukocytosis, with the presence of immature granulocytes in the peripheral blood, making it easily confused with CML. However, white blood cell counts in myelofibrosis are generally lower than those in CML and rarely exceed 30×109/L. The NAP reaction is typically positive. Circulating nucleated red blood cells are persistently observed, with notable abnormalities in red blood cells, such as tear-drop cells. The Ph chromosome and BCR::ABL1 fusion gene are absent. Mutations in genes such as JAK2 V617F, CALR, or MPL may be present. Bone marrow aspiration often results in a dry tap, and reticulin positivity is seen on bone marrow biopsy.
Treatment
The treatment of CML should primarily focus on the early chronic phase, with the goal of preventing disease progression, achieving cytogenetic and molecular remission, and minimizing the risk of transformation. Prognosis is poor once the disease progresses to the accelerated phase or blastic phase.
Treatment of CML in the Chronic Phase (CP)
Management of Hyperleukocytosis
Emergency management of hyperleukocytosis aligns with the protocols outlined in the second section of this chapter, utilizing hydroxyurea and allopurinol. Selected CP patients with extremely high white blood cell counts or symptoms of leukostasis may undergo therapeutic leukapheresis. After a definitive diagnosis, tyrosine kinase inhibitor (TKI) therapy is initiated as the first-line treatment.
Molecular Targeted Therapy
The first-generation TKI imatinib mesylate (IM), a derivative of 2-phenylaminopyrimidine, specifically blocks ATP binding at the ABL1 kinase site, inhibiting tyrosine residue phosphorylation and thus suppressing the proliferation of BCR::ABL1-positive cells. IM treatment achieves a complete cytogenetic remission rate of 92%, with a 10-year overall survival (OS) of up to 84%. Resistance to IM is associated with point mutations in the BCR::ABL1 gene, gene amplification and overexpression, or excessive expression of P-glycoprotein. Inconsistent adherence to therapy can lead to secondary resistance associated with mutations in the BCR::ABL1 kinase domain. Second-generation TKIs, such as nilotinib and dasatinib, achieve a faster deep molecular response (DMR) compared to IM and are becoming viable first-line therapy options. Third-generation TKIs, such as olverembatinib, can overcome the T315I mutation in the ABL1 kinase domain and provide new treatment options for such patients.
During TKI therapy, hematologic toxicities such as leukopenia, thrombocytopenia, and anemia, as well as non-hematologic toxicities such as edema, headache, rash, and elevated bilirubin, may occur. Regular efficacy monitoring is conducted at 3, 6, 12, and 18 months after initiating TKI therapy and thereafter. Patients classified as treatment failures require mutation analysis of the ABL1 kinase domain, with subsequent adjustments to TKI therapy or considerations for hematopoietic stem cell transplantation based on mutation type and drug response. Treatment adherence and close monitoring are essential to achieving optimal therapeutic outcomes. For CP patients who have achieved stable DMR for at least two years, transitioning to TKI-free remission (TFR) is considered a new treatment objective.
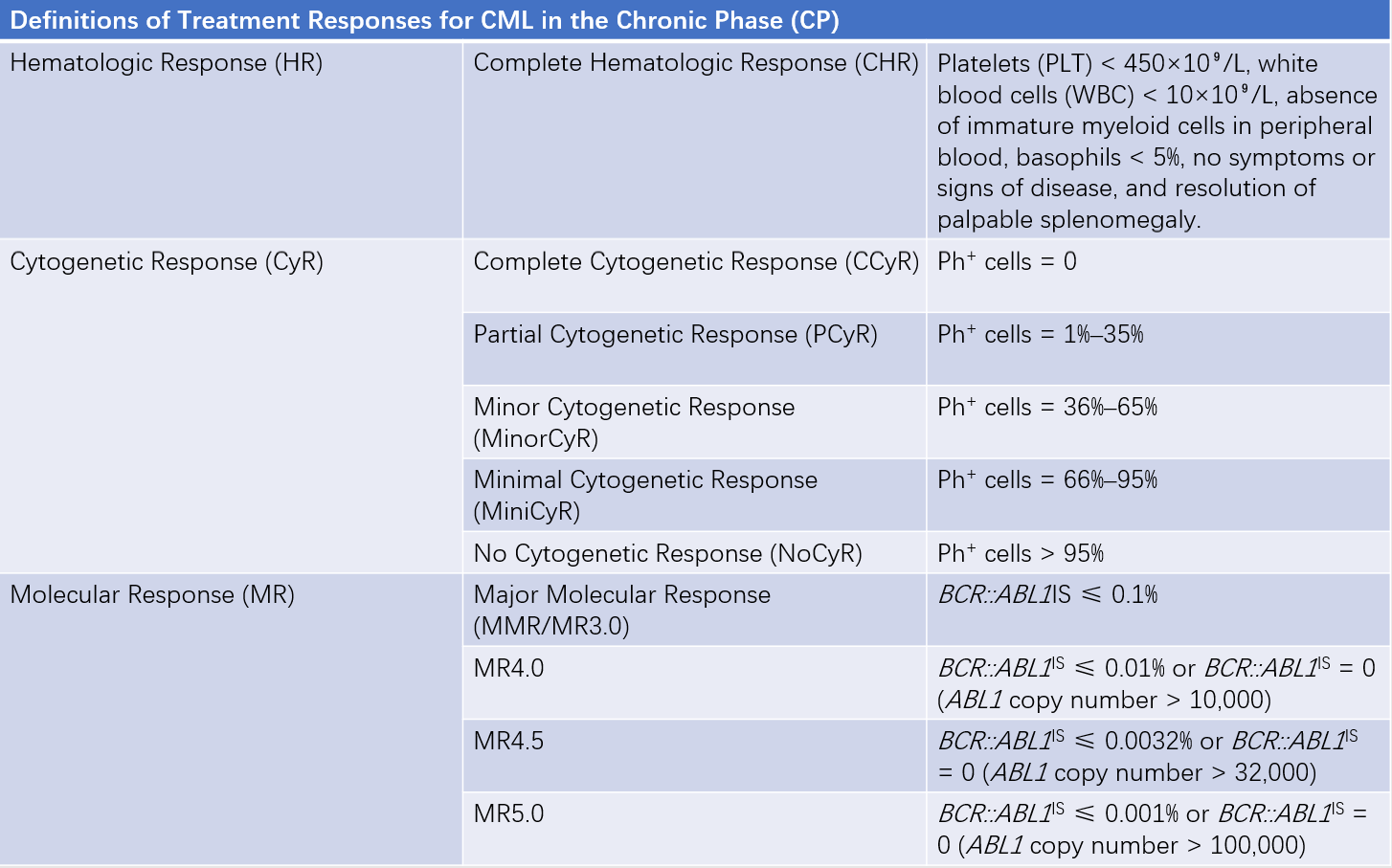
Table 1 Definitions of treatment responses for CML in the chronic phase (CP)
Note: IS refers to the International Scale for standardization.
Interferon
Interferon-α (IFN-α) was the first-line treatment for CML prior to the advent of molecularly targeted therapies. It is now primarily used for patients who are unsuitable for tyrosine kinase inhibitor (TKI) therapy and allogeneic hematopoietic stem cell transplantation (allo-HSCT), as well as for pregnant patients. Given the reproductive toxicity of TKIs, IFN-α, which has limited ability to cross the blood-placenta barrier, is often considered a safer option for patients at any stage of pregnancy.
Other Pharmacological Treatments
Hydroxyurea (hydroxycarbamide, HU)
HU is a cell cycle-specific chemotherapeutic agent noted for its rapid onset of action. White blood cell counts usually begin to decline within two to three days of initiation but increase quickly after discontinuation. Currently, HU monotherapy is reserved for older patients, those with comorbidities, or individuals who are intolerant to both TKIs and IFN-α. It may also be used for rapid leukocyte reduction in cases of leukostasis.
Other Agents
These include cytarabine (Ara-C), homoharringtonine (HHT), arsenic compounds, and busulfan.
Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT)
Allo-HSCT remains an important treatment modality for CML, particularly for patients who are intolerant or resistant to at least two TKIs or those who are in the accelerated phase or blastic crisis. For patients who were in a progressive phase (AP or BC) prior to transplantation, prophylactic TKI treatment may be considered post-transplant.
Treatment of Progressive CML
The accelerated phase (AP) and blastic crisis (BC) are collectively referred to as progressive CML. Upon progression, cytogenetics, molecular levels of BCR::ABL1, and mutational analysis of the BCR::ABL1 kinase domain should be evaluated.
For AP patients who have not previously received TKI treatment, increased doses of first- or second-generation TKIs may be used (e.g., imatinib 600–800 mg/day, nilotinib 800 mg/day, or dasatinib 140 mg/day) with the intent of returning the patient to the chronic phase (CP). In cases where optimal outcomes are not achieved, allo-HSCT is indicated immediately.
For BC patients, chemotherapy in combination with increased doses of TKIs can be administered based on the type of blast phase. This approach seeks to restore CP, enabling allo-HSCT to be performed promptly. The source of stem cells for transplantation is no longer restricted to fully matched donors; haploidentical related donors may also be considered. Post-transplant TKI therapy is recommended to reduce relapse risk, and prophylactic donor lymphocyte infusion (DLI) may be employed to enhance the graft-versus-leukemia effect. For relapse after transplantation, remission may be pursued with DLI ± TKI therapy.
The prognosis for progressive CML remains generally poor, though TKIs have been shown to improve transplant outcomes. Studies have reported a 3-year overall survival (OS) rate of 59% for progressive CML managed with combined TKI and allo-HSCT therapy.
Prognosis
Several factors influence the prognosis of CML, including risk assessments at initial diagnosis (e.g., age, spleen size, peripheral blood platelet count, and percentages of blasts, eosinophils, and basophils in the peripheral blood), the treatment approach (including tolerance to TKIs), and disease progression. Since the introduction of TKIs as first-line therapies, survival has significantly improved for CML patients, with reported 10-year survival rates reaching 85%–90%.