Primary Hodgkin lymphoma (HL) typically originates in the lymph nodes and is characterized by progressive lymph node enlargement. The hallmark pathological feature is the presence of Reed-Sternberg (R-S) cells within a characteristic background of various reactive inflammatory cells, accompanied by varying degrees of fibrosis.
Pathology and Classification
The 2016 WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues divides HL into two major subtypes: Nodular Lymphocyte-Predominant HL (NLPHL) and Classical HL (CHL). NLPHL accounts for approximately 5% of HL cases, while CHL comprises the remaining 95%. Microscopic examination reveals scattered tumor cells—R-S cells and their variants—set against a background of inflammatory cells. Typical R-S cells are large multinucleated or binucleated cells measuring 25–30 μm in diameter, with prominent nucleoli. This is often accompanied by capillary proliferation and varying degrees of fibrosis. The most common CHL subtype is Mixed Cellularity HL (MCHL), followed by Nodular Sclerosis HL (NSHL), Lymphocyte-Rich HL (LRHL), and Lymphocyte-Depleted HL (LDHL).
Nodular Lymphocyte-Predominant HL (NLPHL)
More than 95% of cases show a nodular pattern. Microscopically, this subtype is predominantly composed of small lymphocytes with scattered large tumor cells resembling a "popcorn" appearance. The immunophenotype typically shows a large population of CD20+ small B-cells forming nodules or nodule-like structures. Within these nodules, CD20+ tumor-associated large B-cells are identified, referred to as lymphohistiocytic (L/H) variants of R-S cells. L/H-type R-S cells are CD20+, CD79a+, Bcl-6+, CD45+, and CD75+ in nearly all cases, with approximately half also expressing epithelial membrane antigen (EMA+). Immunoglobulin light and heavy chains are frequently positive. CD15 and CD30 are not expressed.
Classical HL (CHL)
CHL includes:
- Nodular Sclerosis Subtype (NSHL): Comprising 20–40% of CHL cases, the R-S cells often express CD20, CD15, and CD30. Under light microscopy, three defining features are observed: birefringent (Reed-Sternberg) collagen bands separating nodules, nodular lesion patterns, and the presence of "lacunar" R-S cells.
- Lymphocyte-Rich Subtype (LRHL): Characterized by abundant mature lymphocytes with relatively few R-S cells.
- Mixed Cellularity Subtype (MCHL): This subtype features a mix of eosinophils, lymphocytes, plasma cells, and other cell types, with numerous R-S cells often present alongside necrosis. Immunohistochemical staining demonstrates positivity for CD30, CD15, and PAX-5 in tumor cells, and immunoglobulin heavy chain (IgH) or T-cell receptor (TCR) gene rearrangements may be identified.
- Lymphocyte-Depleted Subtype (LDHL): This rare subtype is marked by a significant reduction in lymphocytes and abundant R-S cells, with extensive fibrosis and necrotic lesions often observed.
Clinical Manifestations and Staging
Clinical Features
Lymph Node Enlargement
Painless, progressive enlargement of cervical or supraclavicular lymph nodes is the most common initial symptom, occurring in 60–80% of cases. Axillary lymph node enlargement is also common. Enlarged lymph nodes may remain mobile or become matted and fuse into masses that feel cartilaginous to palpation.
Extranodal Organ Involvement
A small proportion of HL cases involve infiltration of extranodal organs or tissues. Deep lymph node enlargement may cause compression-related symptoms.
Systemic Symptoms
Fever, diaphoresis, pruritus, and weight loss are common systemic symptoms. Persistent unexplained fever is the initial manifestation in 30–40% of HL patients. Such cases often occur in slightly older individuals, predominantly male, with involvement of retroperitoneal lymph nodes. Approximately 1/6 of patients exhibit the characteristic cyclic fever (Pel-Ebstein fever). Localized or generalized pruritus is another feature, more frequently reported in young females. Pruritus may be the only systemic symptom in certain cases.
Other Features
Herpes zoster occurs in 5–16% of HL patients. Lymph node pain following alcohol consumption is a unique but not universal symptom of HL.
Clinical Staging
The Ann Arbor staging system is widely used clinically to classify HL into stages I through IV. Staging is based on the extent of lymph node involvement, with the spleen and Waldeyer's ring each counted as a single lymph node region. Extranodal disease is classified as stage IV, including involvement of the bone marrow, lungs, bones, or liver. This staging system is also used for NHL.
- Stage I: Involvement of a single lymph node region (I) or a single extranodal site (IE).
- Stage II: Involvement of two or more lymph node regions on the same side of the diaphragm (II) or a single confined extranodal organ and its associated lymph nodes, with or without other lymph node regions on the same side of the diaphragm being affected (IIE).
- Note: The number of affected lymph node regions should be specified using a subscript (e.g., II3).
- Stage III: Involvement of lymph node regions on both sides of the diaphragm (III), possibly with involvement of a localized extranodal organ (IIIE), the spleen (IIIS), or both (IIIE+S).
- Stage IV: Diffuse (multifocal) involvement of one or more extranodal organs, with or without associated lymph node enlargement, or isolated extranodal organ involvement with distant (non-regional) lymph node enlargement. Liver or bone marrow involvement, even if limited, is classified as stage IV.
Grouping by systemic symptoms is also used, dividing patients into Group A and Group B. Patients without the following symptoms are designated as Group A, while patients with one or more of these symptoms are categorized as Group B:
- Unexplained fever above 38°C;
- Diaphoresis;
- Weight loss exceeding 10% of body weight within six months.
Involvement sites are recorded using the following symbols: E for extranodal, X for bulky disease (>10 cm in diameter), M for bone marrow, S for spleen, H for liver, O for bone, D for skin, P for pleura, and L for lungs.
Laboratory Tests
Hematological and Bone Marrow Examination
HL commonly features mild to moderate anemia, with eosinophilia present in some patients. Pancytopenia may occur when the bone marrow is extensively infiltrated or when splenomegaly is present. The identification of R-S cells in bone marrow smears confirms bone marrow involvement in HL, and biopsies improve the detection rate.
Radiological and Pathological Examination
The content can be seen in the practices used for non-Hodgkin lymphoma (NHL).
Diagnosis and Differential Diagnosis
The content can be seen in the diagnostic and differential criteria for NHL.
Treatment
Hodgkin lymphoma (HL), though relatively rare, is a highly curable malignancy. Comprehensive treatment primarily involves a combination of chemotherapy and radiotherapy. Early chemotherapy regimens, such as the MOPP regimen, provided complete remission rates of 80%, with a 5-year survival rate of 75% and a long-term disease-free survival rate of 50%. However, a significant proportion of patients experienced secondary malignancies and infertility. The ABVD regimen has shown superior remission rates and 5-year disease-free survival compared to the MOPP regimen and is now considered the first-line chemotherapy protocol for HL. Brentuximab vedotin (BV), a CD30-targeting antibody-drug conjugate, provides greater survival benefits in combination with AVD in advanced stages (III and IV).
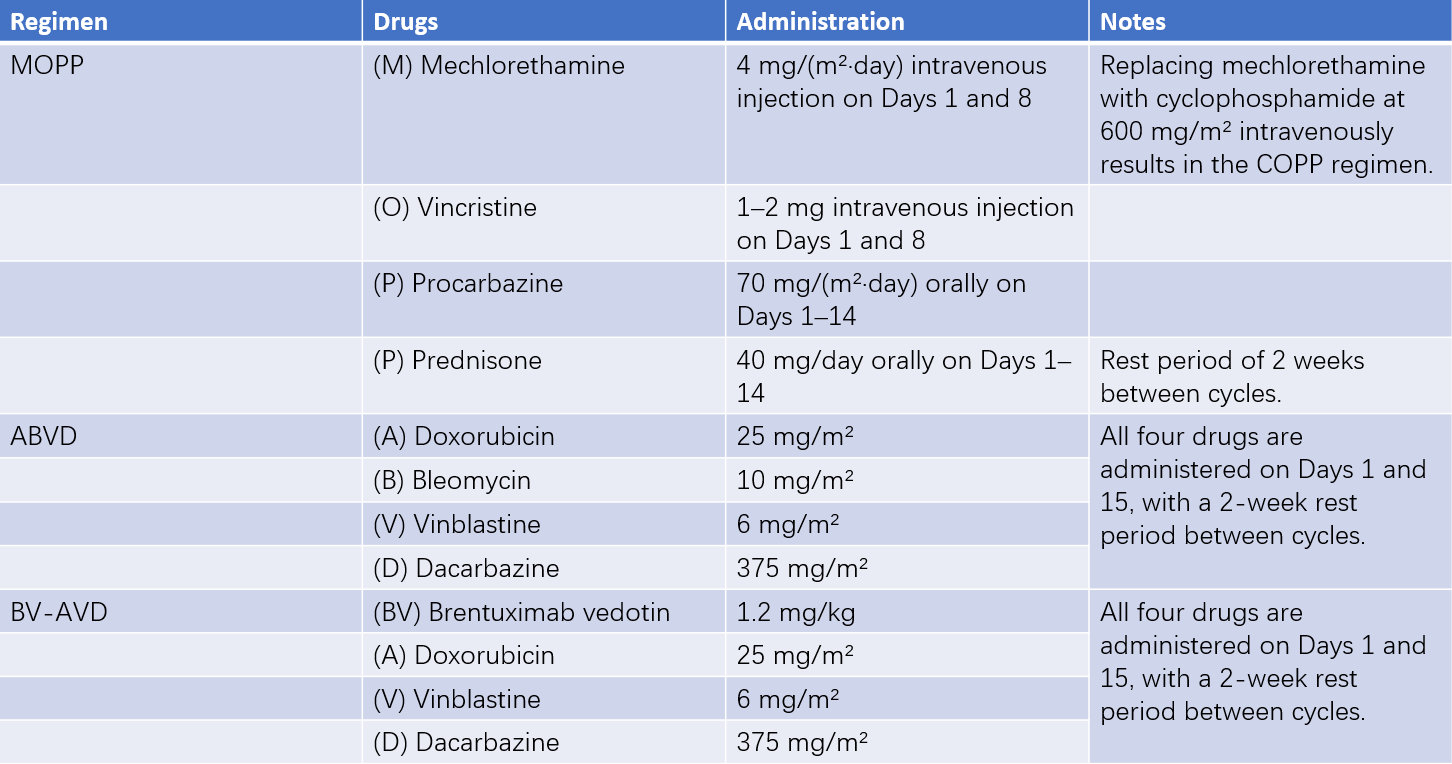
Table 1 Major chemotherapy regimens for Hodgkin lymphoma
Nodular Lymphocyte-Predominant HL (NLPHL)
This subtype is most frequently diagnosed at stage IA and generally has a favorable prognosis. Stage IA may be managed with isolated lymph node excision, observation, or involved-field radiotherapy (20–30 Gy). Treatment of stage II and higher follows the approach for early-stage HL.
Early-Stage (Stage I/II) HL
Systemic chemotherapy is administered, with a trend towards reducing the total dose and field size of radiotherapy. The ABVD regimen is used for chemotherapy. Patients with a favorable prognosis may receive 2–4 cycles of ABVD followed by involved-field radiotherapy (30–40 Gy), while those with an unfavorable prognosis may require 4–6 cycles of ABVD followed by similar radiotherapy.
Advanced-Stage (Stage III/IV) HL
Patients whose disease progresses during chemotherapy or relapses early are considered for salvage high-dose chemotherapy and hematopoietic stem cell transplantation (HSCT). The BV-AVD regimen is currently recommended for this stage. Radiotherapy should be considered for patients with bulky disease prior to chemotherapy or residual tumors after chemotherapy.
Relapsed or Refractory HL (R/R HL)
Patients who relapse after initial radiotherapy may undergo conventional chemotherapy. Resistant or chemotherapy-intolerant cases staged as clinical stage I/II may receive radiotherapy. Patients relapsing after response to standard chemotherapy may undergo second-line chemotherapy or high-dose chemotherapy with autologous HSCT (auto-HSCT). Immunotherapy using PD-1 inhibitors is an option for treating relapsed or refractory classical HL.