Plasma cell dyscrasia refers to a group of disorders caused by the excessive proliferation of clonal plasma cells or immunoglobulin-producing B lymphocytes. This group of diseases is primarily characterized by the presence of excessive monoclonal immunoglobulin or its light chain or heavy chain fragments in the serum or urine.
These disorders include multiple myeloma, monoclonal gammopathy of undetermined significance (MGUS), plasmacytoma (including solitary plasmacytoma and extramedullary plasmacytoma), primary systemic light chain amyloidosis, primary plasma cell leukemia, Waldenström macroglobulinemia, light chain deposition disease, POEMS syndrome, and heavy chain disease. The primary focus of this section is multiple myeloma.
Multiple myeloma (MM) is a disease caused by the clonal proliferation of malignant plasma cells. It is mainly characterized by the malignant proliferation and widespread infiltration of monoclonal plasma cells within the bone marrow, often accompanied by an increase in monoclonal immunoglobulin and/or light chains in the blood or urine. Normal hematopoietic cell proliferation and immunoglobulin secretion are suppressed as a result. Common clinical manifestations include bone pain, anemia, renal dysfunction, hypercalcemia, and infections. MM predominantly affects middle-aged and elderly individuals, with a higher prevalence in males than females. The incidence of MM accounts for approximately 1% of all tumors and represents the second most common hematologic malignancy in many countries.
Etiology and Pathogenesis
The etiology remains unclear. Factors such as genetics, ionizing radiation, chemical exposure, viral infections, and antigenic stimulation may be associated with the development of MM. Although the precise mechanisms are not fully understood, molecular studies suggest that MM is driven by complex genomic alterations and epigenetic abnormalities. Genetic instability is a hallmark of MM, characterized by highly variable chromosomal aberrations. Additionally, interactions between myeloma cells and the bone marrow microenvironment further promote the proliferation of myeloma cells and the development of drug resistance.
Clinical Manifestations
Skeletal Damage
Bone pain is the primary and often the initial symptom of MM, progressively worsening as the disease advances. Pain frequently affects the lower back, sacrum, sternum, and ribs. Pathological fractures may occur due to bone destruction by tumor cells, often accompanied by hypercalcemia. Fractures commonly involve the ribs, clavicle, and thoracic or lumbar vertebrae. The occurrence of bone disease in MM mainly results from an imbalance between osteoclastic and osteoblastic activity.
Anemia
Anemia is another common manifestation of MM. Since it develops gradually, symptoms are often subtle, typically involving mild to moderate anemia. The development of anemia primarily stems from reduced red blood cell production, which is associated with marrow infiltration by myeloma cells, suppression of hematopoiesis, and renal dysfunction.
Renal Dysfunction
Renal damage is a frequent and relatively characteristic clinical manifestation of MM. At diagnosis, some patients present with renal insufficiency or even renal failure. Symptoms may include reduced urine output, foamy urine, and facial or lower extremity edema. Renal dysfunction is closely associated with MM subtypes, with IgD-type being the most common subtype and light chain type following. Renal failure is one of the leading causes of death in MM patients.
Hypercalcemia
Symptoms of hypercalcemia may include loss of appetite, vomiting, fatigue, confusion, polyuria, or constipation. It mostly results from widespread osteolytic lesions and impaired renal function.
Infections
Reduced levels of normal polyclonal immunoglobulins and neutropenia lead to compromised immunity, increasing the susceptibility to various infections. Common infections include bacterial pneumonia and urinary tract infections, and in severe cases, sepsis may occur. Viral infections, with herpes zoster being the most common, are also frequently observed.
Hyperviscosity Syndrome
Symptoms may include dizziness, vertigo, blurred vision, tinnitus, numbness in the fingers, reduced vision, congestive heart failure, altered consciousness, and even coma. Increased levels of M protein in the serum can lead to excessive blood viscosity, resulting in slow blood flow, tissue congestion, and hypoxia. In some patients, the M protein component consists of cryoglobulins, which may cause microcirculatory disturbances, manifested as Raynaud’s phenomenon.
Bleeding Tendency
Frequent manifestations include nosebleeds, gum bleeding, and skin purpura. The mechanisms of bleeding include:
- Thrombocytopenia accompanied by M protein coating the platelet surface, impairing platelet function.
- Coagulation disorders due to M protein binding to fibrin monomers, which interferes with fibrin polymerization; M protein can also directly affect the activity of coagulation factors.
- Vascular wall damage caused by hypergammaglobulinemia and amyloidosis.
Amyloidosis
A minority of patients may develop amyloidosis, often presenting with enlargement of the tongue and parotid glands, myocardial thickening, cardiac enlargement, diarrhea or constipation, lichen-like skin changes, peripheral neuropathy, and liver or kidney dysfunction. Severe cardiac amyloidosis may cause sudden death.
Neurological Impairments
Common symptoms include muscle weakness, numbness, and reduced sensation to pain. Spinal cord compression represents a more severe type of neurological damage. Causes of neurological impairments in MM include infiltration by myeloma cells, mass effects from tumors, hypercalcemia, hyperviscosity syndrome, amyloidosis, deposition of monoclonal light chains, and/or their fragments.
Extramedullary Infiltration
The liver, spleen, lymph nodes, and kidneys are the most commonly affected sites; this is caused by localized infiltration of myeloma cells and the presence of amyloidosis. Liver and spleen enlargement is usually mild. Lymphadenopathy is less common. Other tissues, including the thyroid, adrenal glands, ovaries, testes, lungs, skin, pleura, pericardium, gastrointestinal tract, and central nervous system, may also be involved.
Laboratory and Other Examinations
Blood Cell Analysis
Most patients exhibit varying degrees of normocytic anemia, with a minority displaying hypochromic anemia. On blood smears, red blood cells often appear arranged in a "rouleaux" formation. While white blood cell counts are generally normal, they may sometimes increase or decrease. Platelet counts are usually normal but may occasionally be decreased.
Bone Marrow Examination
Abnormal proliferation of immature or primitive plasma cells is observed in the bone marrow, accounting for more than 10% of cells and reaching as high as 70% to 95% in some cases. Myeloma cells are heterogeneous in size and morphology and tend to aggregate. Binucleated or multinucleated cells may be seen, with 1 to 4 nucleoli visible within the nuclei.
Serum M Protein Detection
The presence of M protein in the serum is a hallmark feature of this disease. Serum protein electrophoresis typically reveals a densely stained monoclonal "M spike" alongside reduced levels of normal immunoglobulins. To identify the M protein subtype, several tests are commonly performed:
- Serum protein electrophoresis.
- Quantification of immunoglobulins.
- Measurement of total and albumin protein levels in the serum.
- Serum immunofixation electrophoresis.
- Quantification of serum free light chains and the kappa-to-lambda free light chain ratio.
Urine Analysis
Urinalysis may appear normal in the early stages for most patients; however, abnormalities are often the first or even the only clinical manifestation in patients with light-chain or IgD-type MM, where renal dysfunction is common. Findings may include proteinuria and casts. Approximately 35% to 65% of MM patients have detectable Bence-Jones proteins (BJP) in the urine. These proteins consist of free light chains, typically either kappa or lambda, excreted by the kidneys.
Biochemical and Serum Protein Analysis
Serum Calcium, Phosphorus, and Alkaline Phosphatase (ALP): Hypercalcemia is common due to widespread bone destruction. Late-stage renal failure may be associated with elevated serum phosphorus. Osteolytic bone lesions generally show normal or slightly elevated ALP levels.
- Serum β2-Microglobulin (β2-M): β2-M is secreted by plasma cells and correlates with tumor burden. Its levels markedly increase with renal dysfunction due to impaired glomerular filtration.
- Serum Total Protein and Albumin: About 95% of patients demonstrate elevated serum total protein, increased globulins, and decreased albumin, which is related to prognosis.
- C-Reactive Protein (CRP) and Lactate Dehydrogenase (LDH): CRP levels reflect disease severity. LDH is related to tumor activity and reflects tumor burden.
- Creatinine (Cr) and Blood Urea Nitrogen (BUN): Both may be elevated in renal impairment.
Cytogenetic Analysis
Current cytogenetic testing methods include chromosomal banding techniques and fluorescence in situ hybridization (FISH). MM typically exhibits complex karyotypic abnormalities involving both numerical and structural chromosomal changes, affecting almost all chromosomes. Certain chromosomal abnormalities are associated with poor prognosis, such as hypodiploidy, del(17p), t(4;14), t(14;16), t(14;20), and 1q amplification.
Imaging Studies
Imaging studies are essential for evaluating bone disease in MM.
- X-Ray Imaging of Bone Lesions: Typical findings include round, well-defined, "punched-out" osteolytic lesions of varying sizes, commonly seen in the skull, pelvis, spine, femur, and humerus.
- Pathologic Fractures: Commonly occur in the spine and ribs.
For many years, X-ray has been widely used for skeletal evaluation in MM because of its low cost, minimal radiation exposure, and simplicity. However, due to its low sensitivity and inability to detect lesions outside the bones, guidelines now recommend higher-sensitivity modalities such as whole-body low-dose CT, whole-body MRI, or PET-CT as the preferred methods for evaluating bone disease in MM.
Diagnostic Criteria, Classification, Staging, and Differential Diagnosis
Diagnostic Criteria
The diagnosis of active plasma cell myeloma (also known as symptomatic myeloma) requires satisfying criterion 1 along with any one of the subcriteria listed under criterion 2.

Table 1 Diagnostic criteria for active (symptomatic) multiple myeloma
Notes: Due to differences in the capacity of clonal plasma cells to synthesize and secrete immunoglobulins, 1–2% of myeloma patients test negative for M-protein. A bone marrow plasma cell proportion ≥10% allows for the diagnosis of "nonsecretory multiple myeloma." Corrected serum calcium (mmol/L) = Total serum calcium (mmol/L) - 0.025 × Albumin concentration (g/L) + 1.0 (mmol/L).
The diagnosis of smoldering plasma cell myeloma (also known as asymptomatic myeloma) requires satisfying criterion 3 in addition to criterion 1 and/or criterion 2.

Table 2 Diagnostic criteria for smoldering multiple myeloma (SMM)
Classification
Multiple myeloma is categorized based on the type of abnormally proliferating immunoglobulin into IgG, IgA, IgD, IgM, IgE, and light chain types, each further divided into kappa and lambda subtypes, resulting in 12 subtypes in total. Additionally, there are biclonal and non-secretory types.
Staging
Staging is performed according to the conventional Durie-Salmon (DS) staging system, the International Staging System (ISS), and the Revised International Staging System (R-ISS). The DS system primarily reflects tumor burden and clinical progression, while the ISS and R-ISS focus on prognosis assessment.
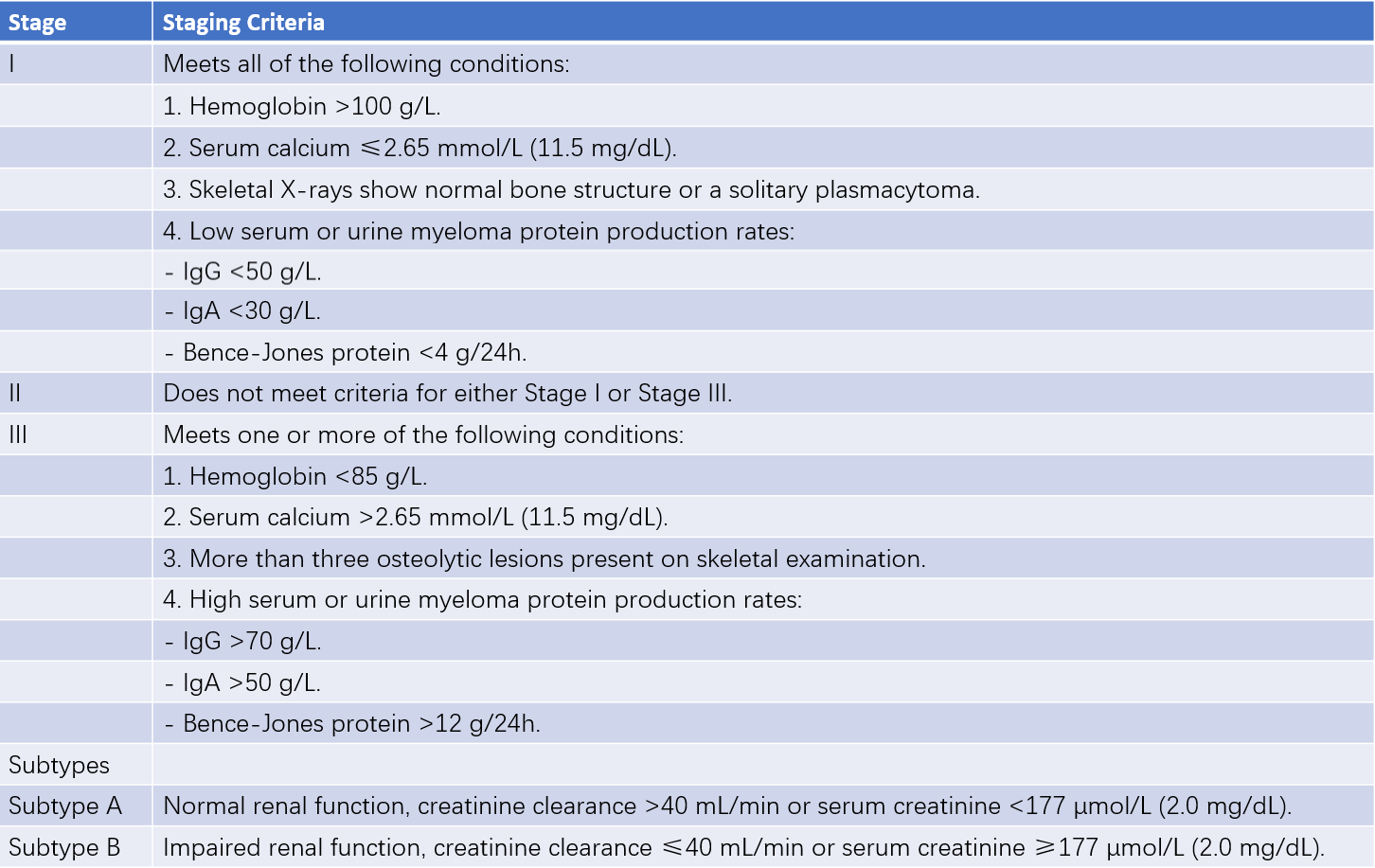
Table 3 Durie-Salmon staging system

Table 4 International staging system (ISS) and revised international staging system (R-ISS)
Notes: High-risk cytogenetics refers to the presence of abnormalities detected by interphase fluorescence in situ hybridization (FISH), including del(17p), t(4;14), or t(14;16).
Differential Diagnosis
MM needs to be differentiated from the following conditions:
Reactive Plasmacytosis
This can be caused by chronic inflammation, typhoid fever, systemic lupus erythematosus (SLE), liver cirrhosis, metastatic cancers, and other conditions. The percentage of plasma cells usually does not exceed 15%, and no morphological abnormalities are observed. The immunophenotype is CD38+, CD56-, with no associated M protein, and IGH gene rearrangement is negative.
Monoclonal Gammopathy of Undetermined Significance (MGUS)
This is characterized by the presence of M protein in the serum and/or urine, along with increased monoclonal plasma cells in the bone marrow. However, the criteria for MM diagnosis are not met, and there is no evidence of end-organ damage related to the condition.
Waldenström Macroglobulinemia (WM)
This is identified by monoclonal IgM in the serum and/or urine and lymphoplasmacytic infiltration in the bone marrow or other tissues. FISH testing generally does not detect IGH translocations such as t(11;14), and molecular testing frequently identifies the MYD88 L265P mutation.
AL Amyloidosis
This is also known as primary systemic light chain amyloidosis, caused by the deposition and degeneration of monoclonal light chains, leading to tissue and organ damage. Congo red staining is positive in biopsy samples.
Diseases Causing Bone Pain and Bone Destruction
Conditions include metastatic bone cancer, senile osteoporosis, renal tubular acidosis, and hyperparathyroidism. These conditions are typically associated with increased osteoblastic activity, often accompanied by elevated serum alkaline phosphatase levels. Identification of the primary lesion or clusters of malignant cells in bone marrow smears can aid in differentiation.
Treatment
Treatment Principles
Systemic therapy, including induction therapy, consolidation therapy (including stem cell transplantation), and maintenance therapy, is recommended for symptomatic MM.
Asymptomatic myeloma is generally not recommended for treatment at this stage.
Treatment of Symptomatic Myeloma
Induction Therapy
The patient's age (generally ≤70 years), physical condition, and comorbidities determine suitability for hematopoietic stem cell transplantation (HSCT).
For transplant-eligible patients, induction regimens should avoid the use of stem cell-toxic drugs such as alkylating agents and nitrosoureas. Regimens containing lenalidomide should ideally be limited to four cycles to prevent failure in stem cell mobilization and collection or delayed hematopoietic recovery. Initial treatments may include the following regimens:
- Bortezomib/Dexamethasone (VD)
- Lenalidomide/Dexamethasone (Rd)
- Lenalidomide/Bortezomib/Dexamethasone (RVD)
- Bortezomib/Doxorubicin/Dexamethasone (PAD)
- Bortezomib/Cyclophosphamide/Dexamethasone (VCD)
- Bortezomib/Thalidomide/Dexamethasone (VTD)
- Thalidomide/Doxorubicin/Dexamethasone (TAD)
- Thalidomide/Cyclophosphamide/Dexamethasone (TCD)
- Lenalidomide/Cyclophosphamide/Dexamethasone (RCD)
For transplant-ineligible patients, in addition to the above regimens, the following options may also be considered:
- Melphalan/Prednisone/Bortezomib (VMP)
- Melphalan/Prednisone/Thalidomide (MPT)
- Daratumumab/Melphalan/Prednisone/Bortezomib (Dara-VMP)
- Daratumumab/Lenalidomide/Dexamethasone (DRd)
Autologous Hematopoietic Stem Cell Transplantation (auto-HSCT)
Renal insufficiency and advanced age are not absolute contraindications for transplantation. Early transplantation is associated with longer event-free survival compared to late-stage transplantation.
Consolidation Therapy
Consolidation therapy is intended to enhance treatment efficacy and deepen the response. Patients who have not achieved maximum therapeutic benefit after induction therapy or auto-HSCT may undergo short-term (2–4 cycles) consolidation therapy with the same regimen used during induction.
Maintenance Therapy
Agents such as lenalidomide, bortezomib, ixazomib, or thalidomide, either as monotherapy or combined with corticosteroids, can be used for maintenance therapy.
Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT)
Young, high-risk, or relapsed/refractory patients may be considered for allo-HSCT.
Supportive Care
Treatment of Bone Disease
Intravenous bisphosphonates or subcutaneous denosumab is recommended for symptomatic MM patients requiring treatment. Bisphosphonates are suitable for all symptomatic patients, while denosumab is preferred for those with renal insufficiency. Surgical intervention may be considered for patients with pathological fractures in long bones, spinal fractures causing spinal cord compression, or spinal instability. For palliative care, low-dose radiation therapy (10–30 Gy) may be utilized to manage uncontrolled pain, imminent pathological fractures, or spinal cord compression.
Management of Hypercalcemia
Hydration and diuresis, combined with the use of denosumab, bisphosphonates, high-dose corticosteroids, and/or calcitonin, are among the management strategies.
Renal Impairment
Hydration and diuretics are used to prevent worsening renal function. Efforts to reduce uric acid production and promote uric acid excretion are recommended. Dialysis is necessary for patients with renal failure. The use of nonsteroidal anti-inflammatory drugs (NSAIDs) and intravenous contrast agents is avoided. Long-term use of bisphosphonates requires monitoring of renal function.
Anemia Management
Erythropoietin (EPO) therapy may be considered.
Infection Management
Intravenous immunoglobulins may be considered in cases of recurrent or life-threatening infections. Prophylaxis against pneumocystis pneumonia and fungal infections is recommended if high-dose dexamethasone is included in the treatment regimen.
Coagulation/Thrombosis
Prophylactic anticoagulation is recommended for patients receiving thalidomide- or lenalidomide-based regimens.
Hyperviscosity Syndrome
Symptomatic patients may undergo plasmapheresis.
Prognosis
The natural course of MM exhibits significant heterogeneity, with wide variations in survival times. MM is still considered an incurable malignancy. However, the application of novel drugs and hematopoietic stem cell transplantation has significantly improved survival outcomes. Median overall survival approaches 10 years in transplant-eligible patients, while it is 4–5 years in non-transplant-eligible patients. Factors influencing prognosis include age, serum LDH levels, renal function, ISS and R-ISS staging, as well as cytogenetic abnormalities.