When blood vessels are damaged, blood can either flow out of the vessel or seep through it. In such cases, the body initiates a series of physiological responses to stop the bleeding, a process known as hemostasis. Hemostasis involves multiple factors and comprises a series of complex physiological and biochemical reactions. Hemorrhagic disorders refer to a group of conditions characterized by spontaneous or excessively prolonged bleeding following minor trauma, resulting from congenital or hereditary factors or acquired defects or abnormalities of hemostatic mechanisms involving blood vessels, platelets, coagulation, anticoagulation, and fibrinolysis.
Normal Hemostatic Mechanisms
Vascular Factors
Vasoconstriction is the body’s earliest physiological response to bleeding. When blood vessels are damaged, local vasoconstriction occurs, narrowing the vessel lumen and reducing or closing the damaged site. Vasoconstriction is regulated by neural reflexes and various mediators.
The endothelium of damaged blood vessels plays the following roles in the hemostatic process:
- Expressing and releasing von Willebrand factor (vWF), which facilitates platelet adhesion and aggregation at the site of injury.
- Expressing and releasing tissue factor (TF), which initiates the extrinsic coagulation pathway.
- Exposing subendothelial collagen, which activates factor XII (FXII) and triggers the intrinsic coagulation pathway.
- Expressing and releasing thrombomodulin (TM), which regulates the anticoagulation system.
Platelet Factors
When blood vessels are injured, platelets participate in hemostasis through adhesion, aggregation, and the release reaction:
- Glycoprotein Ib (GP Ib) on the platelet surface acts as a receptor and, with the bridging action of vWF, adheres platelets to the exposed collagen fibers in the subendothelial space. This forms a platelet plug that mechanically repairs the vessel.
- The glycoprotein IIb/IIIa complex (GP IIb/IIIa) facilitates platelet aggregation by crosslinking with fibrinogen.
- Activated platelets, following aggregation, release a range of active substances such as thromboxane A2 (TXA2) and serotonin (5-HT), further enhancing the hemostatic process.
Coagulation Factors
Damage to vascular endothelium activates both the extrinsic and intrinsic coagulation pathways. In the presence of phospholipids and other factors, a series of enzymatic reactions ultimately result in the formation of a fibrin clot. This clot seals the damaged blood vessel, halting bleeding. Additionally, thrombin and other products from the coagulation cascade play essential roles in promoting blood clot formation and hemostasis.
Coagulation Mechanism
Blood coagulation is a process in which inactive coagulation factors (zymogens) are sequentially and progressively activated through a series of enzymatic reactions, culminating in the conversion of fibrinogen into fibrin. This process is referred to as the "cascade hypothesis" of coagulation. The ultimate outcome of coagulation is the transformation of plasma fibrinogen into fibrin.
Coagulation Factors
Currently, 14 coagulation factors are known to directly participate in the coagulation process. Their names, sites of synthesis, primary biological characteristics, and normal plasma concentrations are summarized in Table 1.
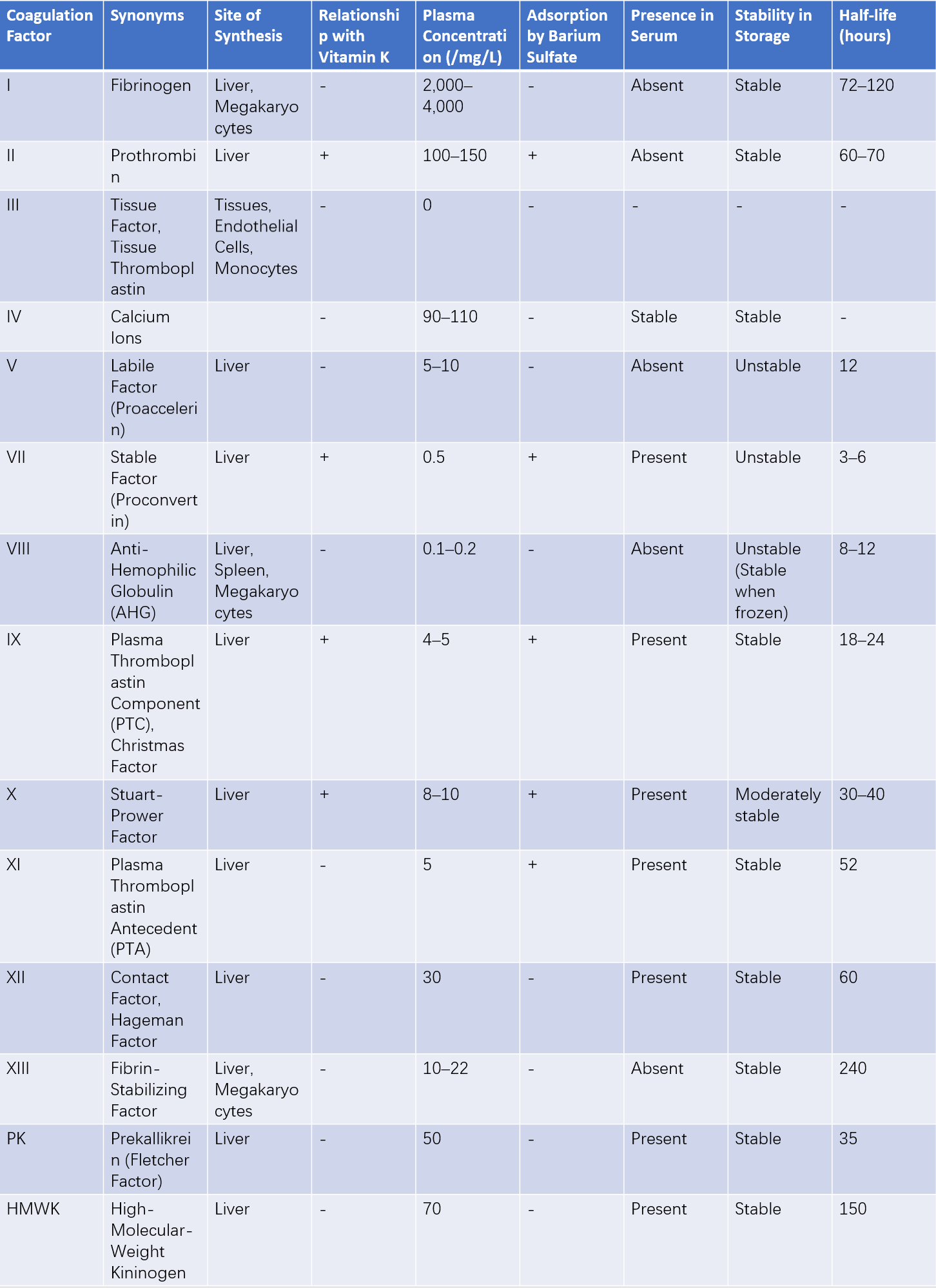
Table 1 Names and characteristics of plasma coagulation factors
Coagulation Pathways
According to the classical coagulation theory, the coagulation process is divided into two pathways based on the initiating event:
- Extrinsic Pathway: Initiated by the contact of blood with TF (also known as the TF pathway).
- Intrinsic Pathway: Initiated by the activation of factor XII (FXII).
Both pathways converge at the activation of factor X (FXa), leading into the common pathway, which eventually results in the formation of fibrin.
Formation of Prothrombinase
Extrinsic Coagulation Pathway
When blood vessels are damaged, endothelial cells express TF, which enters the bloodstream. TF forms a complex with factor VII (FVII) or activated factor VII (FVIIa) in the presence of calcium ions (Ca2+). The TF/FVII or TF/FVIIa complex activates factor X (FX), with the latter being significantly more effective. Additionally, the TF/FVIIa complex can activate factor IX (FIX).
Intrinsic Coagulation Pathway
When blood vessels are damaged, endothelial integrity is disrupted, exposing subendothelial collagen. Factor XII (FXII) comes into contact with negatively charged collagen, leading to its activation and conversion into active factor XII (FXIIa). FXIIa then activates factor XI (FXI), and in the presence of calcium ions (Ca2+), the activated factor XI (FXIa) activates factor IX (FIX). The activated factor IX (FIXa), along with factor VIII (FVIII) and membrane-bound phospholipids (e.g., platelet factor 3, PF3), forms a complex in the presence of Ca2+, which subsequently activates factor X (FX).
After the activation of factor X (FX) by these two pathways, the coagulation process enters the common pathway. In the presence of Ca2+, FXa, factor V (FⅤ), and phospholipids combine to form a complex referred to as prothrombinase.
Thrombin Generation
Prothrombin, an inactive precursor in plasma, is converted into thrombin, a highly proteolytic enzyme, by prothrombinase. The formation of thrombin is a pivotal event in the coagulation cascade, and thrombin performs several additional roles:
- Providing a positive feedback loop to accelerate the conversion of prothrombin into thrombin, a function many times more efficient than prothrombinase itself.
- Inducing irreversible aggregation of platelets, accelerating their activation and release reactions.
- Activating factor XI (FXI).
- Activating factor XIII (FXIII), which facilitates stable fibrin formation.
- Activating plasminogen, thereby enhancing fibrinolysis.
Fibrin Formation
Under the action of thrombin, fibrinogen is sequentially cleaved to release peptides A and B, forming fibrin monomers. These monomers spontaneously polymerize to form unstable fibrin. Subsequently, under the action of activated factor XIII (FXIIIa), stable cross-linked fibrin is formed.
Modern coagulation theory divides the coagulation process into two stages. The first stage is the initiation phase, in which the extrinsic pathway (TF pathway) produces a small amount of thrombin. The second stage is the propagation phase, during which the small amount of thrombin enhances its own generation through positive feedback: activating platelets, facilitating translocation of phosphatidylserine to the outer membrane to enhance phospholipid activity, activating factor V (FⅤ) and factor VIII (FVIII), and activating factor XI (FXI) in the presence of phospholipids and thrombin. This results in the production of sufficient thrombin to complete the normal coagulation process.
Anticoagulation and Fibrinolytic Mechanisms
Beyond the coagulation system, the body also has well-developed anticoagulant and fibrinolytic systems. A dynamic balance is maintained between coagulation and anticoagulation, as well as between fibrin formation and fibrinolysis, ensuring the fluidity of blood.
Composition and Function of the Anticoagulation System
Antithrombin (AT)
Antithrombin (AT) is the most critical anticoagulant in the body, accounting for approximately 75% of the physiological anticoagulant activity in plasma. It is synthesized primarily in the liver and vascular endothelial cells. AT’s main function is to inactivate FXa and thrombin, although it also exhibits some inhibitory activity against other serine proteases such as FIXa, FXIa, and FXIIa. Its anticoagulant activity depends heavily on its interaction with heparin.
Protein C System
The protein C system consists of protein C (PC), protein S (PS), and thrombomodulin (TM). PC and PS are vitamin K-dependent coagulation factors synthesized in the liver. TM is located primarily on the surface of vascular endothelial cells, where it functions as a thrombin receptor. A 1:1 complex forms when thrombin binds to TM, resulting in the cleavage of PC and the generation of activated protein C (APC). APC, with PS as a cofactor, exerts anticoagulant effects by inactivating factor V (FⅤ) and factor VIII (FVIII).
Tissue Factor Pathway Inhibitor (TFPI)
TFPI is a heat-stable glycoprotein, likely produced primarily by endothelial cells. The anticoagulant mechanism of TFPI includes:
- Directly inhibiting FXa.
- Forming a complex with FXa in the presence of Ca²⁺, which subsequently inhibits the TF/FVIIa complex.
Heparin
Heparin is a sulfated glycosaminoglycan synthesized mainly by mast cells in the lung and intestinal mucosa. Its anticoagulant effects include inhibition of FXa and thrombin. These effects are closely associated with AT; heparin binds to AT, inducing a conformational change that exposes AT's active site. The altered AT then forms a 1:1 complex with FXa or thrombin, leading to the inactivation of these serine proteases. Recent studies have shown that low-molecular-weight heparin exhibits a much stronger anti-FXa effect than sodium heparin. Additionally, heparin promotes the release of tissue-type plasminogen activator (t-PA) from endothelial cells, thereby enhancing fibrinolytic activity.
Composition and Activation of the Fibrinolytic System
Composition
The fibrinolytic system primarily consists of plasminogen and its activators, as well as fibrinolysis-related inhibitors.
- Plasminogen (PLG): A single-chain glycoprotein primarily produced in the spleen, eosinophils, and kidneys. Vascular endothelial cells also express plasminogen.
- Tissue-type plasminogen activator (t-PA): The primary plasminogen activator in the human body, synthesized mainly in endothelial cells.
- Urokinase-type plasminogen activator (u-PA): Originally isolated from urine, it is also referred to as urokinase (UK). Its primary forms include pro-urokinase (pro-UK) and the two-chain urokinase-type plasminogen activator.
- Fibrinolysis-related inhibitors: These include α2-antiplasmin (α2-AP), α1-antitrypsin (α1-AP), and α2-antifibrinolysin (α2-AN), among others. They function to inhibit t-PA, plasmin, and other components of the fibrinolytic process.
Activation of the Fibrinolytic System
Intrinsic Pathway
This activation pathway is closely related to the intrinsic coagulation process. When factor XII (FXII) is activated, prekallikrein is converted into kallikrein by the action of FXIIa. Kallikrein then facilitates the conversion of plasminogen into plasmin, initiating the fibrinolytic process.
Extrinsic Pathway
When vascular endothelium or tissue is damaged, t-PA or u-PA is released into the bloodstream, which cleaves plasminogen to generate plasmin, thereby activating the fibrinolytic system.
As a serine protease, plasmin acts on fibrin (and fibrinogen), breaking them down into smaller peptides (A, B, and C) and various fragments collectively referred to as fibrin/fibrinogen degradation products (FDP).
Classification
Hemorrhagic disorders can be classified into several major types based on their etiology and pathogenesis.
Abnormalities of the Vascular Wall
Congenital or Hereditary
- Hereditary hemorrhagic telangiectasia
- Familial simple purpura
- Congenital connective tissue disorders (abnormalities in vessels and their supporting structures)
Acquired
- Infections: Such as sepsis
- Allergic Reactions: Such as Henoch-Schönlein purpura
- Chemical Substances and Drugs: Such as drug-induced purpura
- Nutritional Deficiencies: Such as vitamin C and niacin (vitamin PP) deficiency
- Metabolic and Endocrine Disorders: Such as diabetes mellitus and Cushing's syndrome
- Others: Such as connective tissue diseases, arteriosclerosis, mechanical purpura, and positional purpura.
Platelet Abnormalities
Abnormal Platelet Count
Thrombocytopenia:
- Decreased platelet production (e.g., in aplastic anemia, leukemia, or bone marrow suppression after radiation or chemotherapy)
- Excessive platelet destruction, often associated with immune mechanisms (e.g., primary immune thrombocytopenia [ITP])
- Excessive platelet consumption (e.g., disseminated intravascular coagulation [DIC])
- Abnormal platelet distribution (e.g., hypersplenism)
Thrombocytosis (with platelet dysfunction):
Abnormal Platelet Quality
Congenital or Hereditary: Glanzmann's thrombasthenia, Bernard-Soulier syndrome, and other syndromes involving large platelets.
Acquired: Caused by antiplatelet drugs, infections, uremia, abnormal gammopathies, and other conditions. Acquired platelet dysfunction is more common.
Coagulation Abnormalities
Congenital or Hereditary
- Hemophilia A and B
- Hereditary factor XI deficiency
- Rare hereditary coagulation disorders, including deficiencies of prothrombin, and factors V, VII, and X, among others.
Acquired
- Coagulopathies associated with liver diseases
- Vitamin K deficiency
- Uremic coagulation abnormalities
Abnormalities in Anticoagulation and Fibrinolysis
Primarily acquired conditions:
- Excessive use of heparin
- Overdose of coumarin drugs or poisoning with rodenticide (warfarin-based)
- Increased levels of immune-related anticoagulants
- Envenomation from animal toxins
- Overdose of thrombolytic agents
Combined Hemostatic Mechanism Abnormalities
Congenital or Hereditary:
- von Willebrand disease (vWD)
Acquired:
- Disseminated intravascular coagulation (DIC)
Diagnosis
The patient’s medical history and clinical manifestations often provide clues to the cause and diagnosis of bleeding.
Medical History
Bleeding Characteristics
Key factors include the age at which bleeding begins, the location, duration, volume of bleeding, whether there has been umbilical cord bleeding at birth or delayed bleeding, and whether there is recurrent bleeding at the same site. Generally, bleeding into the skin or mucosa (e.g., petechiae or purpura) is often associated with vascular or platelet abnormalities. Deep hematomas or joint bleeding raise concerns about coagulation disorders. Delayed bleeding after trauma or surgery often suggests abnormalities in fibrinolysis.
Triggers for Bleeding
The occurrence of spontaneous bleeding or its association with surgery, trauma, or exposure to medications is considered.
Underlying Diseases
Relevant conditions include liver diseases, kidney diseases, gastrointestinal disorders, diabetes, immune disorders, and certain specific infections.
Family History
A history of similar bleeding disorders or diseases among paternal, maternal, or close relatives is explored.
Other Considerations
Diet, nutritional status, occupation, and environmental factors are reviewed, and in female patients, menstrual history is assessed, especially for cases of menorrhagia, prolonged menstrual bleeding, or the passage of large blood clots.
Physical Examination
Signs of Bleeding
These include the range and location of bleeding, the presence of deep bleeding such as hematomas, oozing at wound sites, and whether the distribution is symmetric or asymmetric.
Signs of Related Diseases
Signs to be noted include pallor (indicative of anemia), hepatosplenomegaly, lymphadenopathy, jaundice, spider angiomas, ascites, edema, joint deformities, or visible abnormal clusters of dilated capillaries on the skin.
General Signs
These include heart rate, respiratory rate, blood pressure, and peripheral circulation status.
The diagnostic significance of patient history and physical examination for hemorrhagic disorders is summarized in Table 2.
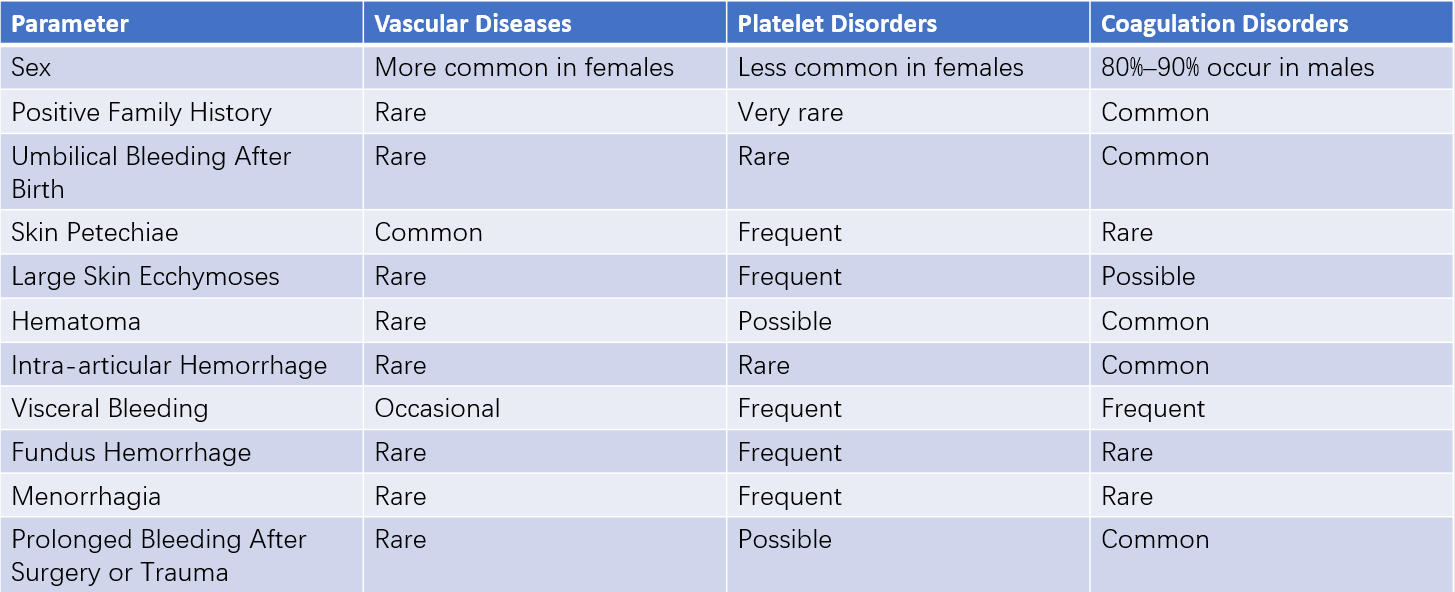
Table 2 Clinical differentiation of common hemorrhagic disorders
Laboratory Tests
The clinical features of hemorrhagic disorders are generally suggestive but rarely definitive. Most bleeding disorders require laboratory tests for confirmation of diagnosis. Laboratory investigations typically follow a sequence of screening, confirmation, and specialized tests.
Screening Tests
Simple screening tests help provide a general idea of the location and mechanism of hemostasis disorders.
- Vascular or Platelet Abnormalities: Including bleeding time (BT), platelet count, and morphological examination.
- Coagulation Abnormalities: Including activated partial thromboplastin time (APTT), prothrombin time (PT), thrombin time (TT), and fibrinogen (Fbg) levels.
Confirmatory Tests
The sensitivity and specificity of screening tests are relatively poor, and some bleeding disorders may exhibit normal screening results. Conversely, abnormal screening test results may be caused by other factors (e.g., severe liver dysfunction, uremia, or use of anticoagulant medications). When screening tests are abnormal and clinical suspicion of a bleeding disorder exists, specialized or more precise laboratory tests are performed to confirm the diagnosis.
- Vascular Abnormalities: Tests for von Willebrand factor (vWF), endothelin-1 (ET-1), and thrombomodulin (TM).
- Platelet Abnormalities: Tests include platelet count and morphology, platelet aggregation studies, surface expression of P-selectin (CD62P), and solid-phase detection of direct platelet antigens (GPIIb/IIIa and GPIb/IX) using monoclonal antibodies. The International Society on Thrombosis and Haemostasis (ISTH) recommends flow cytometry to assess inherited and acquired platelet quantity and functional disorders.
- Coagulation Abnormalities: Functional and activity assays for coagulation factors can clarify plasma levels.
- First Phase of Coagulation: Assessing antigens and activities of FXII, FXI, FX, FIX, FVIII, FVII, FV, and tissue factor (FTF).
- Second Phase of Coagulation: Measuring prothrombin antigens and activities.
- Third Phase of Coagulation: Assessing fibrinogen, abnormal fibrinogen, fibrin monomers, and FXIII antigens and activities.
- Anticoagulant Abnormalities:
- Measuring antithrombin (AT) antigens and activity, or thrombin-antithrombin complexes (TAT).
- Tests for protein C (PC), protein S (PS), and thrombomodulin (TM).
- Detection of FVIII inhibitors.
- Assessing lupus anticoagulants or antiphospholipid antibodies.
- Fibrinolysis Abnormalities:
- Tests include protamine sulfate precipitation test (3P test), fibrin degradation product (FDP) measurement, and D-dimer determination.
- Plasminogen determination.
- Tests for t-PA, plasminogen activator inhibitor (PAI), and measurement of plasmin-antiplasmin complexes (PIC).
The significance of common coagulation and bleeding tests in the diagnosis of hemorrhagic disorders is summarized in Table 3.
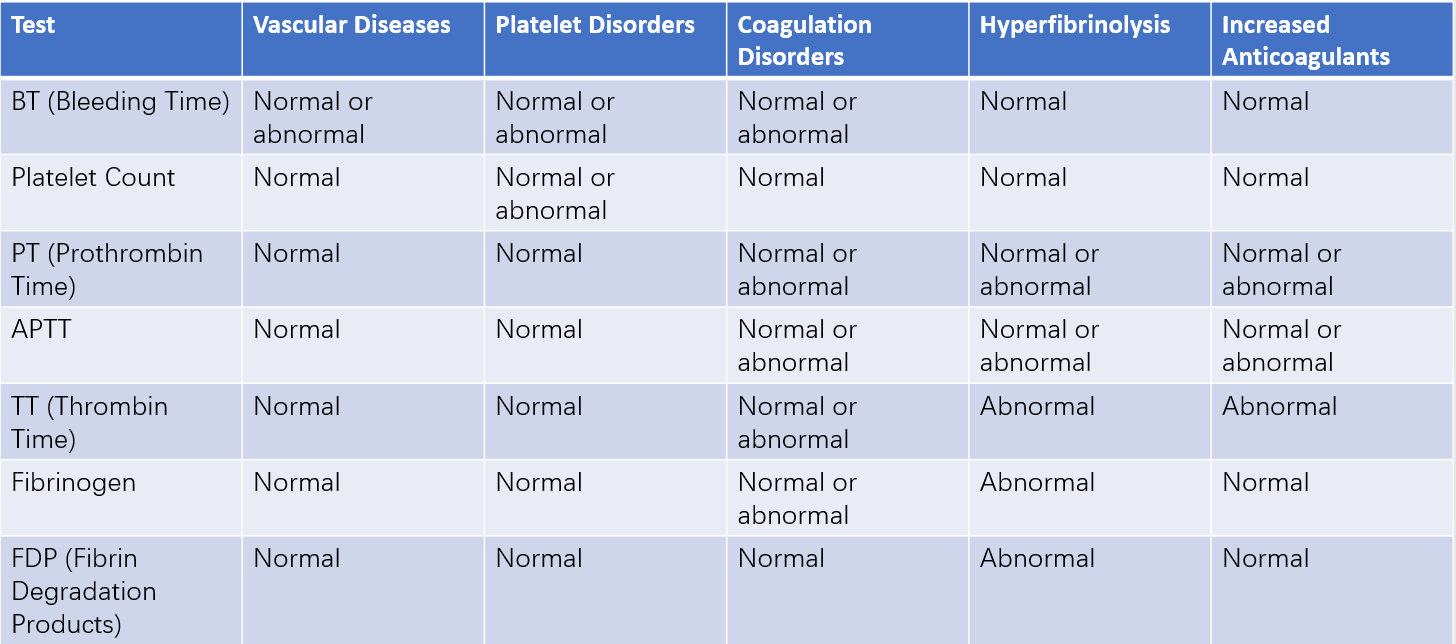
Table 3 Significance of common bleeding and coagulation tests in the diagnosis of hemorrhagic disorders
Diagnostic Process
The diagnostic process progresses methodically, beginning with common disorders before considering rare diseases, moving from simple to complex, and employing common tests before advanced ones.
- Determining whether a bleeding disorder is present.
- Distinguishing between vascular abnormalities, platelet abnormalities, coagulation disorders, or other conditions.
- Identifying whether the issue is due to a quantitative abnormality or a functional defect.
- Using the medical history, family history, and specific tests to preliminarily determine whether the condition is congenital, hereditary, or acquired.
- In cases of congenital or hereditary disorders, conducting genetic and molecular biological testing to ascertain the exact etiology and pathogenesis.
Prevention and Treatment
Prevention and Management of Underlying Causes
This approach primarily applies to acquired hemorrhagic disorders.
Management of Underlying Diseases
This includes controlling infections, actively treating liver and biliary diseases, and suppressing abnormal immune responses.
Avoidance of Substances and Medications That May Aggravate Bleeding
For conditions such as vascular hemophilia and platelet function disorders, medications like aspirin, indomethacin, and ticlopidine that impair platelet function should be avoided. For bleeding disorders caused by coagulation dysfunction, such as hemophilia, caution should be applied with the use of anticoagulants, including warfarin and heparin.
Hemostatic Treatments
Supplementation of Platelets and/or Coagulation Factors
In emergency situations, transfusion of fresh plasma or fresh frozen plasma serves as a reliable approach for supplementation or replacement, as these contain all coagulation factors except TF and Ca²⁺. Additionally, platelet concentrate, fibrinogen, prothrombin complex concentrate, cryoprecipitate, and factor VIII can be supplemented based on the patient’s condition.
Hemostatic Drugs
Several categories of hemostatic drugs are commonly used in clinical practice:
- Drugs that constrict blood vessels, increase capillary density, and improve vascular permeability: Examples include carbazochrome, troxerutin, vasopressin, vitamin C, and glucocorticoids.
- Drugs required for the synthesis of coagulation-related components: Such as vitamin K.
- Antifibrinolytic drugs: Examples include epsilon-aminocaproic acid (EACA) and para-aminomethylbenzoic acid (PAMBA).
- Drugs that promote the release of hemostatic factors: Such as desmopressin.
- Recombinant activated factor VII (rFVIIa): rFVIIa is a new coagulation agent. It acts directly or forms a complex with tissue factor to promote the activation of factor X and the formation of thrombin.
- Topical hemostatic agents: Examples include thrombin, batroxobin, and absorbable gelatin sponges.
Drugs That Stimulate Platelet Production
Various cytokines regulate the proliferation, differentiation, and platelet production by megakaryocytes at different stages. Currently available clinical drugs in this category include thrombopoietin (TPO) and interleukin-11 (IL-11).
Local Management
Local measures include applying pressure bandages, immobilization, and surgical ligation of local blood vessels.
Other Treatments
These approaches are primarily applicable to acquired hemorrhagic disorders.
Immunotherapy
Immunotherapy can be used for hemorrhagic disorders related to immune factors, such as immune thrombocytopenia (ITP), severe hemophilia A and B with high-titer inhibitors, and similar conditions. Treatment options include glucocorticoids and anti-CD20 monoclonal antibodies.
Plasma Exchange
For diseases such as thrombotic thrombocytopenic purpura (TTP), plasma exchange can be used to remove antibodies or related pathogenic factors.
Surgical Treatment
Surgical options include splenectomy, evacuation of hematomas, joint arthroplasty, and joint replacement.
Gene Therapy
Gene therapy holds the potential to bring new hope to patients with hereditary hemorrhagic disorders.