Hemophilia is a group of hereditary coagulation disorders caused by deficiencies in prothrombin activator formation, primarily including hemophilia A and hemophilia B, with hemophilia A being more prevalent. The condition is characterized by a positive family history, onset in childhood, uncontrolled bleeding after minor trauma or spontaneously, hematoma formation, and joint bleeding.
Etiology
Hemophilia A, also known as hereditary factor VIII (FVIII) deficiency, is a common hereditary bleeding disorder. FVIII circulates in a complex with von Willebrand factor (vWF). Activated FVIII participates in the intrinsic activation of factor X, while vWF functions as an adhesive molecule facilitating platelet adhesion to damaged vascular endothelium and serves to stabilize and protect FVIII. The FVIII gene is located on the long arm of the X chromosome (Xq28). Defects in this gene due to inheritance or mutation result in insufficient synthesis of FVIII, leading to impaired intrinsic coagulation and a bleeding tendency.
Hemophilia B, also known as hereditary factor IX (FIX) deficiency, involves a single-chain glycoprotein that, when activated by factor XIa, participates in the intrinsic activation of factor X. The FIX gene is located on the long arm of the X chromosome (Xq27). When defects are caused by inheritance or mutation, insufficient FIX production occurs, resulting in similar coagulation impairments and a bleeding tendency.
Genetic Inheritance
Both hemophilia A and B are inherited in an X-linked recessive manner.
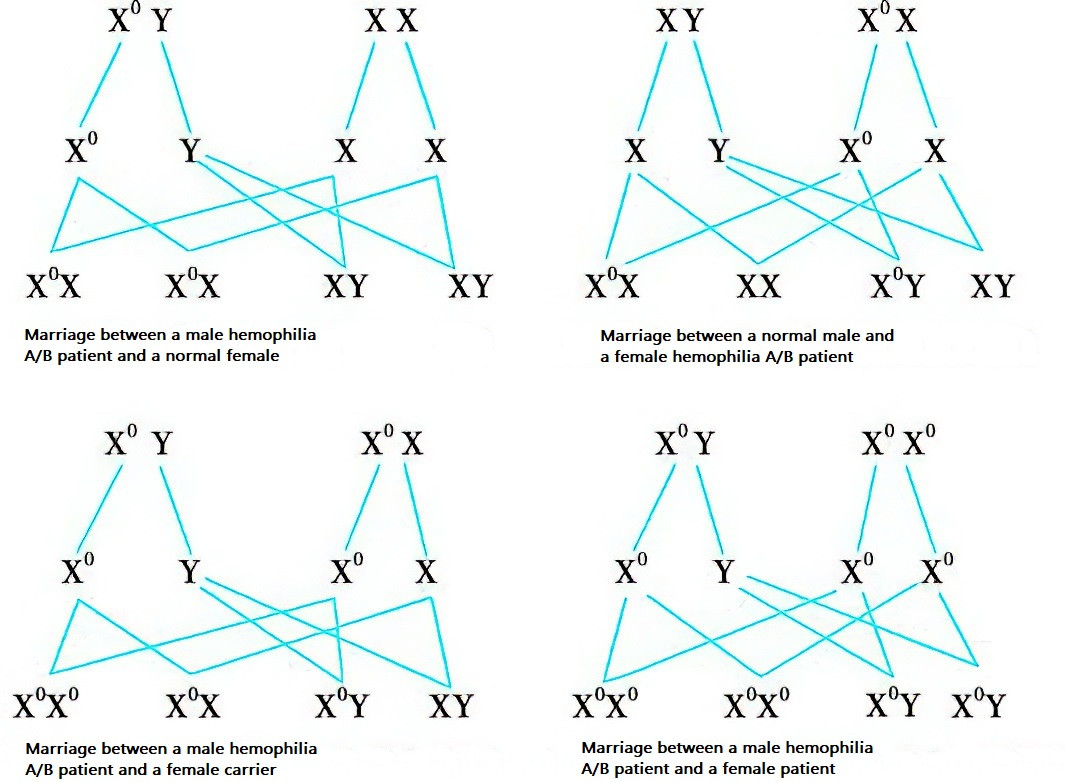
Figure 1 Patterns of genetic inheritance for hemophilia A and B
XY: Normal male;
XX: Normal female;
X0Y: Male patient with hemophilia A/B;
X0X: Female carrier of hemophilia A/B;
X0X0: Female patient with hemophilia A/B.
Clinical Features
Bleeding
The severity of bleeding correlates with the extent of coagulation factor deficiency. Based on coagulation factor activity levels in plasma, hemophilia can be classified into three types:
- Severe: Factor activity <1%.
- Moderate: Factor activity of 1–5%.
- Mild: Factor activity of 5–40%.
Bleeding in hemophilia is often spontaneous or prolonged following minor trauma or small surgical procedures (e.g., tooth extraction, tonsillectomy) and exhibits the following characteristics:
- Congenital in onset and lifelong in duration.
- Frequently hematomas in soft tissues or deep muscles.
- Recurrent bleeding in weight-bearing joints such as the knees and ankles, possibly leading to joint swelling, stiffness, deformities, osteoporosis, joint ossification, and associated muscle atrophy.
Symptoms and Signs Due to Hematoma Compression
Hematomas compressing surrounding nerves may result in localized pain, numbness, or muscle atrophy. Compression of blood vessels may cause ischemic necrosis, congestion, or edema in the affected area. Ureteral compression can lead to urinary obstruction. Bleeding in the floor of the mouth, posterior pharyngeal wall, larynx, or neck may cause respiratory distress or even asphyxiation. Intestinal wall bleeding may result in intussusception or obstruction.
Complications of Hemophilia
Complications of hemophilia can be categorized into two types. The first includes complications arising from acute or recurrent bleeding, such as hemophilic arthropathy, pseudotumor formation, or compartment syndrome. The second type involves complications from replacement therapy, including transfusion-related viral hepatitis, HIV infection, and the development of inhibitors against FVIII/FIX.
Laboratory Tests
Screening Tests
Platelet count, prothrombin time (PT), thrombin time (TT), and fibrinogen levels are normal. Activated partial thromboplastin time (APTT) is prolonged, although it may be only slightly prolonged or normal in mild cases of hemophilia.
Diagnostic Tests
Intrinsic coagulation factor activities associated with prolonged APTT are measured. FVIII activity (FVIII:C) combined with FVIII antigen (FVIII:Ag) assays confirm hemophilia A, while FIX activity combined with FIX antigen assays confirm hemophilia B. Additional tests include APTT correction tests, vWF:Ag measurements, and, if necessary, inhibitor testing, to differentiate hemophilia from acquired coagulation factor deficiencies and von Willebrand disease.
Genetic Testing
Gene analysis of the patient and family members provides confirmation of the causative mutation. This facilitates carrier detection and allows for prenatal diagnosis within the same family.
Diagnosis and Differential Diagnosis
Diagnostic Reference Criteria
Male patients, with or without a family history, are common; those with a family history often show patterns consistent with X-linked recessive inheritance. Female patients are rare.
Bleeding can occur at any site, most commonly in joints, muscles, and deep tissues, and can be spontaneous or triggered by mild trauma or minor surgical procedures. Hematoma formation and joint deformity are frequent findings.
Differential Diagnosis
Hemophilia requires differentiation from von Willebrand disease, other hereditary coagulation factor deficiencies, and acquired coagulation factor deficiencies.
Treatment and Prevention
The treatment approach primarily involves comprehensive management centered on replacement therapy. Key principles include:
- Strengthening self-protection to minimize trauma and bleeding.
- Prompt and effective management of bleeding episodes to prevent complications from occurring or progressing.
- Avoidance of aspirin, non-steroidal anti-inflammatory drugs, and other medications that may interfere with platelet aggregation.
- Regular follow-up in a multidisciplinary hemophilia treatment center and implementation of home-based care.
Preventive therapy is recommended for patients with severe hemophilia.
Genetic counseling and prenatal diagnosis to reduce the incidence of hemophilia.
Replacement Therapy
Replacement therapy remains the cornerstone of hemophilia treatment, involving supplementation with the deficient clotting factors, either factor VIII (FVIII) or factor IX (FIX). This approach is critical for both the prevention and management of bleeding. Based on the treatment objectives, replacement therapy can be classified into on-demand therapy and prophylaxis. Common therapeutic products include plasma-derived and recombinant clotting factor concentrates, fresh frozen plasma, cryoprecipitates, and prothrombin complex concentrates. Recent advances in technology and treatment paradigms have also introduced non-factor therapeutic agents, such as bispecific antibodies targeting factor IXa and factor X, and antibodies inhibiting tissue factor pathway inhibitors (TFPI).
The half-life of FVIII is 8–12 hours and that of FIX is 18–24 hours. Therefore, FVIII replacement requires continuous intravenous infusion or twice-daily administrations, whereas FIX can be administered once daily.
The dosage of FVIII or FIX can be calculated as follows:
FVIII dosage (IU): Body weight (kg) × Desired increase in activity level (%) ÷ 2
FIX dosage (IU): Body weight (kg) × Desired increase in activity level (%)
To achieve hemostasis, FVIII:C or FIX:C levels must exceed 20%. For severe bleeding or planned moderate-to-major surgery, activity levels of FVIII or FIX should exceed 40%.
Prophylactic therapy involves the regular administration of hemostatic agents (clotting factors or non-factor agents) to enhance coagulation and effectively prevent bleeding in patients with hemophilia. Prophylactic regimens should be tailored based on factors such as patient age, clinical severity, past bleeding history, treatment goals, presence of inhibitors, and the pharmacokinetics of the medication.
General Treatment
Initial management of joint bleeding often follows the RICE principle: rest and immobilization of the affected limb, ice application, compression to the affected area, and elevation of the limb.
Home-Based Therapy
Home-based therapy for hemophilia has been widely practiced. Except for patients with anti-FVIII antibodies, unstable disease conditions, or children under three years old, most patients can manage their condition at home. Patients and their families should receive education on the pathophysiology, diagnosis, and treatment of hemophilia. Home therapy should initially be conducted under the guidance of professional physicians. Education should also cover injection techniques, hematology, orthopedic care, psychological well-being, physical therapy, and prevention of HIV and viral hepatitis.
Management of Complications
For recurrent bleeding unresponsive to medical treatment or musculoskeletal complications due to lack of access to preventive therapy, surgical treatment can be considered after coagulation function is corrected.
For hemophilia patients with inhibitors, the management strategy depends on the level of the inhibitor and the severity of bleeding. The main approaches include immune suppression therapy and bypassing agents to reduce bleeding and eliminate inhibitors.
Gene Therapy
Gene therapy involves the delivery of normal FVIII or FIX genes into the patient’s body using vectors, allowing the patient to produce functional FVIII or FIX with significantly elevated factor levels. Several gene therapy products have now entered clinical use, although their high costs remain a limiting factor.