A pituitary tumor refers to a group of neoplasms originating from the pituitary gland and embryonic craniopharyngeal epithelial cells, accounting for 10%-20% of intracranial tumors. Most pituitary tumors are benign. These tumors may manifest as excessive or insufficient secretion of pituitary hormones or as symptoms caused by local tumor compression. In some cases, no clinical manifestations are observed.
Etiology and Classification
Based on endocrine functionality, pituitary tumors are primarily categorized into two types: functional and non-functional tumors.
Functional pituitary tumors include prolactinomas (PRL tumors), growth hormone-secreting tumors (GH tumors), adrenocorticotropic hormone-secreting tumors (ACTH tumors), thyroid-stimulating hormone-secreting tumors (TSH tumors), luteinizing hormone/follicle-stimulating hormone-secreting tumors (LH/FSH tumors), and mixed tumors.
Non-functional pituitary tumors do not secrete biologically active hormones but may synthesize and release hormone fragments (e.g., α-subunits).
Among these types, PRL tumors, GH tumors, and non-functional pituitary tumors are the most common.
Pituitary tumors are also classified according to their size: microadenomas (<10 mm in diameter) and macroadenomas (≥10 mm in diameter).
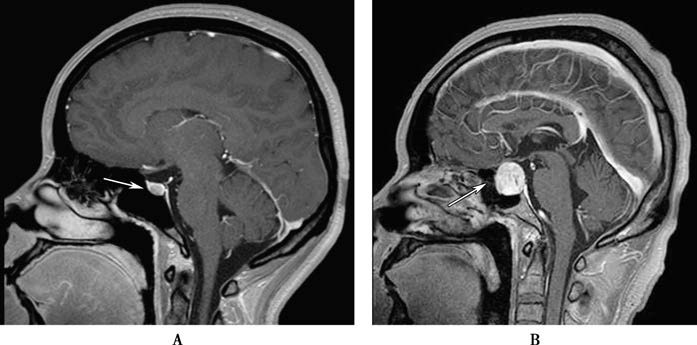
Figure 1 Pituitary tumor (pituitary MRI)
A. Pituitary microadenoma; B. Pituitary macroadenoma
Based on tumor growth patterns, pituitary tumors can be divided into expansive and invasive types.
According to the site of origin, these tumors can be further classified into primary tumors and metastatic tumors (primary cancers of the breast, lungs, or gastrointestinal tract, for example, may metastasize to the pituitary).
Pathogenesis
The development of pituitary tumors is potentially linked to intrinsic cellular defects within the pituitary and regulatory dysfunction of the hypothalamus. A mutation in a pituitary cell can result in the activation of oncogenes and/or inactivation of tumor suppressor genes, leading to hypothalamic regulatory abnormalities. Under the influence of intrinsic and extrinsic factors, monoclonal mutated cells proliferate continuously, ultimately forming a pituitary tumor.
Some cases exhibit familial patterns. Specific gene mutations, including MEN1 (multiple endocrine neoplasia type 1), Gsα (G-protein-regulatory subunit), and AIP (aryl hydrocarbon receptor-interacting protein), may lead to the development of one or more pituitary adenomas. Mutations in the MEN1 gene result in loss of function, commonly causing tumors in the parathyroid gland, pancreatic islets, and pituitary. Mutations in GNAS (the gene encoding Gsα) activate adenylate cyclase, promoting tumor cell division and excessive secretion of growth hormone. Mutations in the AIP gene are associated with familial pituitary adenomas, often resulting in GH tumors or mixed GH-PRL tumors.
Clinical Manifestations
Hormone Secretion Abnormalities
Pituitary tumors may cause clinical syndromes due to excessive hormone secretion. Conversely, tumor enlargement may compress normal pituitary tissue or the pituitary stalk, reducing the secretion of corresponding pituitary hormones. The latter may lead to secondary hypogonadism, adrenal cortical insufficiency, hypothyroidism, or growth hormone deficiency. However, these conditions often present with mild and insidious symptoms. Clinical manifestations of excessive or deficient pituitary hormone secretion are discussed in detail in relevant chapters.
Mass Effect
Headache
Headache may occur when the tumor compresses the diaphragma sellae. Persistent headache can result from pressure on pain-sensitive structures, such as the walls of large blood vessels. When the tumor breaks through the diaphragma sellae, headache may actually reduce. Additionally, compression and traction on the dura mater by the tumor may lead to midbrain aqueduct obstruction, resulting in symptoms such as headache, nausea, and vomiting.
Cranial Nerve Compression
Upward growth of the tumor into the suprasellar region may compress the optic chiasm, optic nerves, or optic tracts. Compression of the optic chiasm may cause visual field defects, such as bitemporal hemianopia. Compression of the optic nerve or optic tract may lead to optic atrophy or vision loss. Obstruction of retinal venous return may result in papilledema. Anterior growth of the tumor may compress the olfactory nerve or olfactory tract, resulting in loss of the sense of smell. Lateral extension of the tumor into the cavernous sinus can result in cavernous sinus syndrome, involving damage to the oculomotor, trochlear, abducens, and trigeminal nerves. This can manifest as dilated pupils, loss of light reflex, ptosis, diplopia, restricted eye movement, sensory deficits in the first and second divisions of the trigeminal nerve, and loss of the corneal reflex.
Invasion of Other Brain Tissues
Invasion of the frontal or temporal lobes by the tumor may lead to seizure-like convulsions, hemiplegia, corticospinal tract signs, and psychiatric symptoms. Hypothalamic involvement can result in diabetes insipidus, hypersomnia, thermoregulatory disturbances, and autonomic nervous system dysfunction. Tumor invasion into the skull base or sphenoid sinus may cause cerebrospinal fluid rhinorrhea.
Pituitary Apoplexy
Hemorrhage within the tumor can lead to pituitary apoplexy, presenting with severe headache, nausea, vomiting, hyponatremia, sudden vision loss, extraocular muscle paralysis, stupor, coma, meningeal irritation, and increased intracranial pressure. In severe cases, a pituitary crisis can occur. MRI or CT imaging of the pituitary can confirm the diagnosis.
Diagnosis and Differential Diagnosis
A detailed medical history and physical examination (such as neurological assessment, fundoscopic evaluation, visual acuity, and visual field tests) provide important clues for the diagnosis of pituitary tumors. Functional diagnosis involves pituitary hormone testing and functional tests. Imaging diagnosis primarily relies on MRI, as it can identify microadenomas smaller than 3 mm in diameter and reveal adjacent structural abnormalities as well as areas of tumor invasion. The final diagnosis depends on pathological examination and immunohistochemical analysis.
Diseases to be differentiated from pituitary tumors include Rathke's cleft cysts, craniopharyngiomas, lymphocytic hypophysitis, optic nerve gliomas, ectopic pinealomas, internal carotid aneurysms, retrobulbar optic neuritis, meningiomas, empty sella syndrome, suprasellar germinomas, and metastatic pituitary carcinomas, among others.
Treatment
The main objectives are:
- Correction of hormone secretion disorders.
- Alleviation of mass effects caused by the tumor.
- Preservation of pituitary structure and function as much as possible.
- Prevention and management of tumor recurrence.
- Reduction of complications.
Surgical Treatment
Surgery is generally the first choice for most pituitary tumors, except for prolactinomas (PRL tumors). The most common surgical approach is transsphenoidal surgery, which accesses the sella turcica through anatomical pathways of the nasal cavity and sinuses. The cure rate for this approach is approximately 70%-80%, with a recurrence rate of 5%-15%. Postoperative complications include transient diabetes insipidus, cerebrospinal fluid rhinorrhea, local hematoma or abscess formation, low rates of infection, and a mortality rate of less than 1%. For large macroadenomas that have invaded the suprasellar or parasellar regions, a transcranial surgical approach via the frontal route is often necessary. However, this approach typically has a lower cure rate and a higher risk of postoperative complications.
Medical Treatment
Medications can be considered for functional tumors. Bromocriptine is commonly used for prolactinomas and growth hormone-secreting tumors (GH tumors). ACTH-secreting tumors may also respond to medical treatment. Somatostatin analogs are frequently used for GH tumors and may also be considered for TSH tumors and LH/FSH tumors. Specific drug regimens are detailed in the chapters covering different types of pituitary tumors.
Radiation Therapy
Radiation therapy is primarily used as an adjunct to surgery. Indications include significant residual tumor following surgery that is not well-controlled by medication, patient refusal of surgery, or tumor recurrence. The effectiveness of radiation depends on factors such as the radiation dose and the operator’s expertise in performing the treatment. Radiation methods include conventional radiotherapy, three-dimensional conformal/stereotactic radiotherapy, and proton beam therapy. Post-radiation complications frequently include hypopituitarism, particularly deficiencies of GH, LH/FSH, and ACTH. Other complications, such as optic neuritis, visual impairment, and brain atrophy, may also occur. Clinical selection of radiation therapy requires careful consideration.