Hypopituitarism, also referred to as anterior pituitary hypofunction, is a clinical syndrome caused by insufficient secretion of one or more anterior pituitary hormones due to damage to the hypothalamus, the hypothalamic-pituitary axis, or the pituitary gland. The prevalence is reported to be between 29–45.5 cases per 100,000, with an incidence of approximately 42 cases per 1,000,000. It can occur in both men and women. About 50% of patients exhibit deficiencies in three or more anterior pituitary hormones. In women during the perinatal period, hypopituitarism resulting from ischemic necrosis of the anterior pituitary gland is known as Sheehan syndrome.
Hypopituitarism originating from primary pituitary lesions is classified as primary hypopituitarism, while that caused by hypothalamic or hypothalamic-pituitary axis lesions is classified as secondary hypopituitarism. Based on the number of affected pituitary hormones, it can be further divided into panhypopituitarism (complete hormonal deficiency), partial hypopituitarism (deficiency of multiple hormones), and isolated hypopituitarism (deficiency of a single hormone).
Etiology and Pathogenesis
The causes of hypopituitarism include acquired or genetic factors (such as pituitary tumors, parasellar tumors, pituitary ischemic necrosis, traumatic brain injury, sellar surgery, radiation therapy, infiltrative diseases, infections, pituitary inflammation, and pituitary apoplexy) as well as idiopathic cases where the underlying cause is unknown.

Table 1 Causes of hypopituitarism
Pituitary and Perisellar Tumors
Pituitary tumors are the most common cause of hypopituitarism. The mechanisms include destruction of normal pituitary tissue or compression of the gland by the tumor, impairment of the pituitary stalk leading to disrupted blood supply or transportation of hypothalamic-releasing hormones, and tumor-associated hemorrhage resulting in pituitary apoplexy. Most patients with large pituitary adenomas experience deficiencies in one or more pituitary hormones, with growth hormone (GH), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) being the most commonly affected. Tumors near the sellar region, such as craniopharyngiomas, germ cell tumors, meningiomas, gliomas, and hamartomas, may also compress the pituitary and result in hypopituitarism.
Pituitary Ischemic Necrosis
During the perinatal period, factors such as placenta previa, retained placenta, or uterine atony may cause massive hemorrhage and shock, leading to ischemic necrosis and fibrosis of the anterior pituitary gland, resulting in hypopituitarism (Sheehan syndrome). The anterior pituitary lacks a direct arterial blood supply and relies on a dense capillary network from the hypothalamic-pituitary portal system. This makes it vulnerable to ischemia without an established collateral circulation. Additionally, hypertrophy of the anterior pituitary during pregnancy increases susceptibility to ischemic damage. The posterior pituitary’s blood supply is independent of the portal system and hence is typically unaffected by peripartum hemorrhage.
Sellar Surgery or Trauma
Pituitary tumor removal surgery frequently leads to hypopituitarism. Severe traumatic brain injuries can cause hemorrhage, necrosis, and fibrosis of the hypothalamus or pituitary gland. Injury to the pituitary stalk may disrupt the connection between the hypothalamus and the portal system or damage the portal system itself, leading to ischemic infarction of the anterior pituitary gland and resulting in partial or complete hypopituitarism, often accompanied by impaired posterior pituitary function.
Radiation-Induced Damage
Radiation therapy to the sellar area or systemic radiation therapy can cause hypopituitarism. Radiation-related damage often occurs years after treatment. For this reason, patients who receive radiation therapy require regular evaluations of pituitary gland function. Radiation damage typically spares the posterior pituitary.
Infections and Infiltrative Diseases
Conditions such as tuberculosis, syphilis, and fungal infections can result in damage to the hypothalamic-pituitary axis. Other infiltrative diseases, including sarcoidosis, histiocytosis, Wilson’s disease, and hemochromatosis, may also invade the hypothalamic-pituitary region, leading to hypopituitarism. Sarcoidosis and histiocytosis are often accompanied by diabetes insipidus.
Empty Sella Syndrome
Empty sella syndrome occurs when a congenital weakness in the diaphragma sellae allows the arachnoid membrane to herniate into the sella turcica. It may also develop secondary to pituitary infarction. This condition can compress or displace the pituitary gland. If more than 90% of pituitary tissue is compressed or atrophied, hypopituitarism may result.
Pituitary Inflammation
Lymphocytic hypophysitis is the most common form of pituitary inflammation. It involves diffuse lymphocytic and plasma cell infiltration of the anterior pituitary gland and predominantly affects women. It typically manifests during pregnancy or shortly after delivery and is often accompanied by mild increases in prolactin (PRL) levels and elevated erythrocyte sedimentation rates. Other autoimmune diseases are commonly coexistent.
Autoimmune hypophysitis may present as isolated anterior pituitary hormone deficiency, partial deficiency, or complete anterior pituitary hormone deficiency. Its imaging findings can resemble those of a pituitary tumor, making differentiation on MRI or CT challenging, and it is often misdiagnosed as a pituitary tumor. In necessary cases, a biopsy may be performed. Less common forms of hypophysitis include granulomatous, xanthomatous, plasmacytic, or IgG4-related hypophysitis.
Pituitary Apoplexy
The most common cause of pituitary apoplexy is sudden hemorrhage within a pituitary tumor, but it may also occur in individuals with postpartum hemorrhage, diabetes, hypertension, or shock. Sudden tumor enlargement can compress normal pituitary tissue and adjacent neural structures, presenting with acute sellar region compression syndrome and/or meningeal irritation signs accompanied by hypopituitarism. The onset is rapid and often includes symptoms such as headache, nausea, vomiting, hyponatremia, and in severe cases, pituitary crisis. A definitive diagnosis can be made through MRI or CT.
Genetic Causes
These are rare. Congenital anterior pituitary hypoplasia, such as those caused by mutations in the POU1F1 and PROP1 genes, can impair the development of GH-, PRL-, and TSH-secreting cells, leading to deficiencies of these hormones. Mutations in the HESX1 gene may also result in multiple pituitary hormone deficiencies and may involve underdevelopment of the diaphragma sellae and optic nerve tracts. Additionally, congenital hypothalamic dysfunction, as seen in Kallmann syndrome or Prader-Willi syndrome, may also be associated with hypopituitarism.
Clinical Manifestations
The onset of hypopituitarism is insidious, with symptoms that are highly variable and nonspecific. Common symptoms include fatigue, loss of appetite, amenorrhea, hyponatremia, hypoglycemia, and hypotension. In some cases, patients may exhibit no noticeable clinical symptoms, and the condition can only be diagnosed through hormone measurements or functional tests. Hypopituitarism may also have an acute onset with life-threatening severity. The clinical manifestations depend on the degree and type of pituitary hormone deficiency, as well as the extent of atrophy of the corresponding target glands. In cases of hypopituitarism caused by pituitary tumors or radiation, deficiencies in GH and FSH/LH often appear first, followed by deficiencies in TSH and ACTH.
LH and FSH Deficiency Syndrome
Deficiency of LH and FSH leads to hypogonadism, which is the most common manifestation of hypopituitarism. In women, symptoms may include lactation failure postpartum, amenorrhea, breast atrophy, reduced or lost libido, decreased vaginal lubrication, dyspareunia, infertility, and loss of pubic or axillary hair. In men, symptoms may include decreased libido, erectile dysfunction, sparse facial, pubic, and axillary hair, testicular atrophy, reduced muscle mass, and increased fat deposition. Serum levels of estradiol or testosterone are low, but FSH and LH levels are not correspondingly elevated.
GH Deficiency Syndrome
In children, GH deficiency primarily manifests as growth retardation, while in adults, it causes reduced muscle mass and strength, decreased endurance, central obesity, impaired concentration and memory, dyslipidemia, and osteoporosis. Due to the nonspecific nature of symptoms, it is often overlooked.
TSH Deficiency Syndrome
TSH deficiency results in secondary hypothyroidism, with clinical features similar to those of primary hypothyroidism. In secondary hypothyroidism, serum levels of thyroid hormones (FT3 and FT4) are reduced, but TSH levels are reduced or normal. In primary hypothyroidism, TSH levels are elevated.
ACTH Deficiency Syndrome
ACTH deficiency leads to secondary adrenal insufficiency, which presents with similar features to primary chronic adrenal insufficiency (refer to Chapter 7 of this section). In both conditions, plasma cortisol levels are often <140 nmol/L (5 µg/dL). However, in ACTH deficiency, plasma ACTH levels are reduced (or normal), resulting in hypopigmentation, pallor, and reduced pigmentation of the areolae, in contrast to primary adrenal insufficiency, where ACTH levels are elevated, leading to hyperpigmentation.
Compression Syndrome of the Pituitary and Surrounding Structures
Compression of the pituitary or adjacent structures may produce symptoms such as headache and visual impairment. In some cases, symptoms and signs of elevated intracranial pressure may be observed. If the hypothalamus is involved, clinical manifestations related to hypothalamic syndromes, such as anorexia nervosa or thermoregulatory disturbances, may occur.
Pituitary crisis is considered a medical emergency. In cases of panhypopituitarism, various stressors such as infections, sepsis, diarrhea, emesis, dehydration, starvation, cold exposure, acute myocardial infarction, stroke, surgery, trauma, anesthesia, or the use of sedatives, hypnotics, or hypoglycemic agents may trigger a pituitary crisis. This condition may present with life-threatening symptoms, including hyperpyrexia (>40°C), hypothermia (<35°C), hypoglycemia, hyponatremia, hypotension (shock), altered mental status, delirium, seizures, or coma.
Diagnosis
The onset of hypopituitarism is gradual, with symptoms being nonspecific, which makes it prone to misdiagnosis. Unexplained fatigue, loss of appetite, amenorrhea, hyponatremia, hypoglycemia, or hypotension (or shock) should raise suspicion of this condition. A history of massive hemorrhage and shock during delivery, along with postpartum lack of lactation and amenorrhea, is valuable for diagnosing Sheehan syndrome. In suspected cases, timely evaluation of anterior pituitary function is warranted.
The diagnosis is primarily based on a thorough medical history, clinical presentation, hormonal level assessments, and functional tests of the anterior pituitary. Reduced target gland hormones (cortisol, thyroid hormones, estradiol, or testosterone) accompanied by low or normal levels of corresponding pituitary hormones (ACTH, TSH, FSH/LH) indicate hypopituitarism. In postmenopausal women, low FSH/LH levels also suggest this condition. For mild cases, functional tests of the anterior pituitary (e.g., insulin-induced hypoglycemia test, ACTH stimulation test, or GnRH stimulation test) may assist in the diagnosis. Imaging studies are recommended if necessary, with MRI being the preferred method.
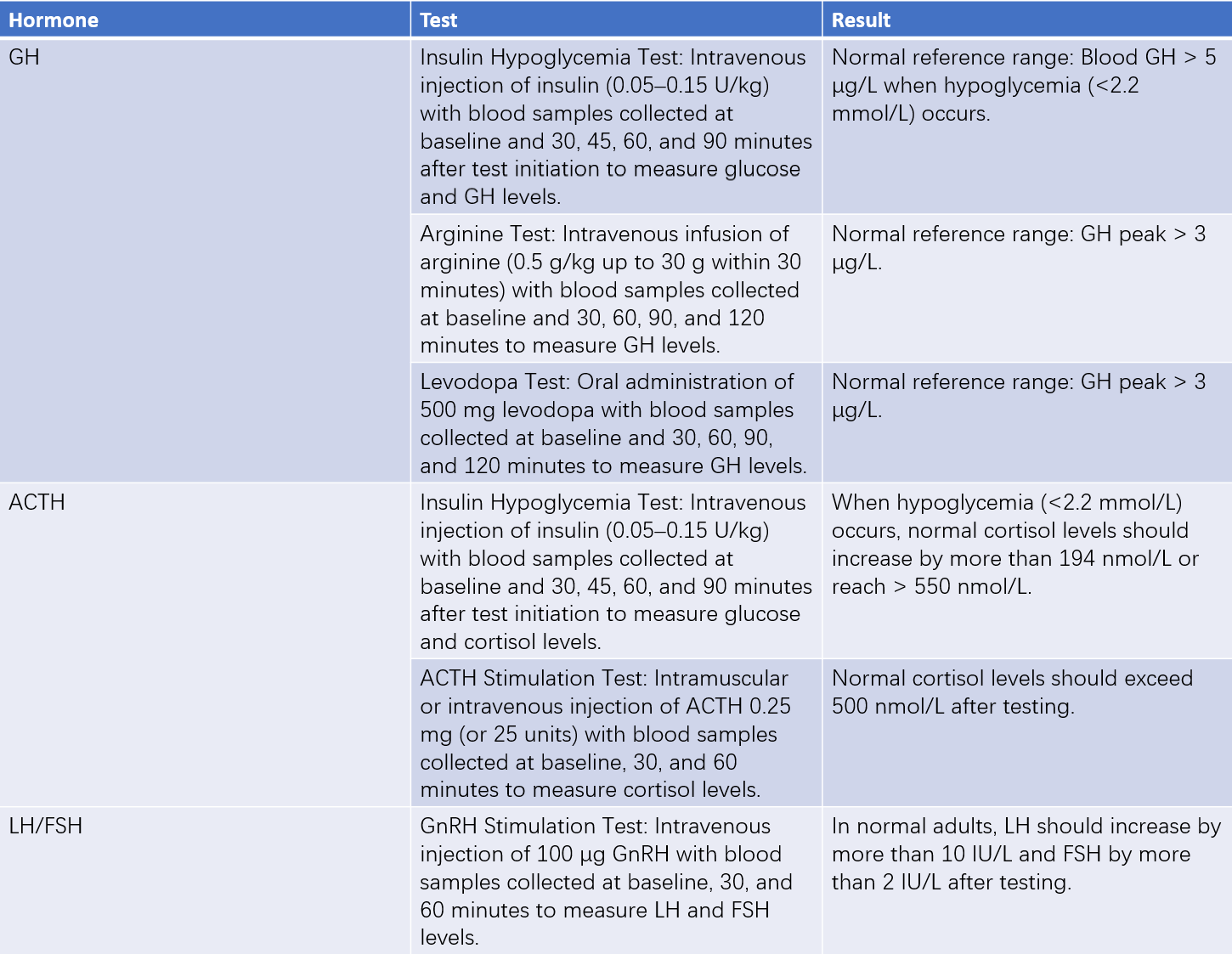
Table 2 Tests related to hypopituitarism
It is worth noting that patients taking glucocorticoids may also exhibit reduced plasma cortisol and ACTH levels. It is important to carefully review the medication history and differentiate based on physical findings, such as the presence of Cushingoid features (e.g., moon face, plethoric appearance).
Treatment
Treatment involves addressing the underlying cause and providing hormone replacement therapy. Hormone replacement therapy aims to mimic physiological conditions as closely as possible, improving symptoms while avoiding overtreatment.
Treatment of the Underlying Cause
Hypopituitarism can result from various causes, and treatment should target the underlying etiology. Surgical, radiotherapeutic, or chemotherapeutic approaches may be used for tumors. In lymphocytic hypophysitis, glucocorticoids are an option. For sellar mass lesions, alleviation of compression and destruction as well as relief of symptoms of intracranial hypertension remain critical. Patients should minimize exposure to infections, overexertion, and stressors.
Hormone Replacement Therapy
Treatment of Growth Hormone Deficiency
Growth hormone supplementation can improve quality of life and reduce complications. Dosage should be individualized, as there is no universally accepted standard for replacement. However, concerns remain regarding the potential for long-term growth hormone therapy to increase tumor risk or recurrence, and its high cost has limited its widespread use. Further research is needed to evaluate the long-term benefits in patients with hypopituitarism.
Treatment of Gonadotropin Deficiency
For individuals without fertility needs, sex hormone replacement is an appropriate treatment. Estrogen and progestin replacement therapy in women can restore menstruation and improve quality of life, though hormone supplementation is typically recommended to stop after menopause. In men, testosterone replacement therapy is an option. For patients desiring fertility, options include gonadotropin replacement therapy or pulsatile GnRH therapy.
Treatment of TSH Deficiency
Secondary hypothyroidism is managed with thyroid hormone replacement therapy, as with primary hypothyroidism. The monitoring of serum TSH levels is not useful for evaluating therapeutic efficacy in secondary hypothyroidism; adjustments in dosage should be based on serum thyroid hormone levels (e.g., FT3, FT4). In cases with concurrent ACTH and TSH deficiencies, glucocorticoid replacement should precede thyroid hormone supplementation, as isolated thyroid hormone replacement can exacerbate clinical manifestations of ACTH deficiency.
Treatment of ACTH Deficiency
Upon confirmation of ACTH deficiency, physiological doses of adrenal corticosteroids should be initiated promptly. The replacement dosage of glucocorticoids is determined based on clinical circumstances. Typically, this involves 10–20 mg of hydrocortisone daily (e.g., 10 mg in the morning, 5 mg at noon, and 5 mg in the evening, with a daily maximum of 30 mg) or prednisone (e.g., 5 mg in the morning and 2.5 mg in the afternoon). Regular follow-up evaluations are necessary during treatment, with adjustments made based on the patient’s condition, body weight, blood pressure, blood glucose, and blood lipids. The measurement of plasma ACTH, cortisol, or urinary free cortisol is not useful for evaluating treatment efficacy.
Management of Pituitary Crisis
If pituitary crisis is suspected, treatment should begin immediately, and blood samples should be drawn before therapy to measure relevant hormone levels.
Management during a crisis includes:
- Correction of Hypoglycemia: Immediate intravenous injection of 40–80 ml of 50% glucose solution, followed by continuous intravenous infusion of 5% glucose saline solution.
- Adequate Glucocorticoid Administration: Hydrocortisone (200–300 mg/day) or dexamethasone (5–10 mg/day) should be added to intravenous fluids and administered in divided doses.
- Correction of Dehydration and Electrolyte Disturbances: Intravenous infusion of 5% glucose saline solution can manage electrolyte imbalances. For severe hyponatremia, higher-concentration saline solutions may be necessary.
- Correction of Shock: Blood pressure typically improves with the aforementioned treatments. In severe cases where blood pressure does not respond adequately, vasopressors and comprehensive anti-shock interventions are required.
- Other Measures: Infection, often the most common trigger, requires appropriate antibiotic therapy based on the patient's condition. For hypothermia, warming with hot water bottles or electric blankets ensures body temperature rises above 35°C, followed by small doses of thyroid hormone after glucocorticoid treatment. For hyperpyrexia, physical and chemical cooling can be used. Sedatives should be used with caution.