Idiopathic pulmonary arterial hypertension (IPAH) refers to pulmonary arterial hypertension (PAH) of unknown cause. It was previously known as primary pulmonary arterial hypertension. Pathologically, it is characterized by plexogenic pulmonary arteriopathy, which includes concentric or eccentric intimal proliferation, medial hypertrophy, plexiform lesions, and necrotizing arteritis.
Epidemiology
European data estimate the prevalence of PAH in adults to be at least 15 cases per million population, with an incidence of at least 2.4 cases per million population per year. The prevalence of IPAH is estimated to be at least 5.9 cases per million population. According to the first National Institutes of Health (NIH) registry study in the United States in 1981, the average age of patients with IPAH was 36 years. In recent years, more older individuals have been diagnosed with IPAH, with recent studies reporting an average age of 50-65 years.
Etiology and Pathogenesis
The etiology of IPAH remains unknown. Current evidence suggests that its development may be related to genetic factors, autoimmunity, endothelial dysfunction, and smooth muscle dysfunction in the pulmonary vasculature.
Genetic Factors
Between 11% and 40% of sporadic IPAH cases exhibit mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene. Some cases also involve mutations in the activin receptor-like kinase 1 (ACVRL1) gene, endoglin, or SMAD9.
Immune and Inflammatory Responses
Immune regulation may play a role in the pathological process of IPAH. Approximately 29% of IPAH patients have significantly elevated antinuclear antibody levels, although they lack specific antibodies associated with connective tissue diseases. Macrophages, T lymphocytes, and B lymphocytes have been observed infiltrating plexiform lesions in IPAH, suggesting that inflammatory cells contribute to the disease's onset and progression.
Endothelial Dysfunction in the Pulmonary Vasculature
Pulmonary vascular tone is regulated by a balance of vasoconstrictors and vasodilators secreted by the endothelium. Key vasoconstrictors include thromboxane A2 (TXA2) and endothelin-1 (ET-1), while vasodilators include prostacyclin and nitric oxide (NO). An imbalance in the expression of these factors leads to smooth muscle contraction and pulmonary arterial hypertension.
Potassium Channel Defects in Vascular Smooth Muscle Cells
Proliferation and hypertrophy of vascular smooth muscle are observed, along with dysfunction of voltage-gated potassium (Kv) channels. Reduced potassium (K+) efflux results in membrane depolarization, allowing calcium ions (Ca2+) to enter the cells, which leads to vasoconstriction.
Clinical Manifestations
Symptoms
The symptoms of IPAH are non-specific. In the early stages, patients are often asymptomatic, experiencing discomfort only during intense physical activity. As pulmonary arterial pressure increases, systemic symptoms gradually develop.
Dyspnea is the most common symptom and is often the initial complaint. It typically presents as exertional dyspnea that progressively worsens, eventually occurring even at rest. This symptom is related to reduced cardiac output and ventilation-perfusion mismatch.
Chest pain is caused by myocardial ischemia due to increased right ventricular afterload, increased oxygen consumption, and reduced coronary blood flow. It often occurs during physical activity or emotional stress.
Dizziness or syncope results from a sudden reduction in cerebral blood flow due to decreased cardiac output. They commonly occur during activity but may also happen at rest.
Hemoptysis is usually mild, though massive hemoptysis can occasionally occur and may be fatal.
Other symptoms include fatigue and weakness, which are often overlooked. About 10% of patients exhibit Raynaud's phenomenon. Enlarged pulmonary arteries may compress the recurrent laryngeal nerve, causing hoarseness (Ortner syndrome).
Signs
The physical signs of IPAH are associated with pulmonary arterial hypertension and increased right ventricular load.
Auxiliary Examinations
Blood Tests
Hemoglobin levels may be elevated as a compensatory response to chronic hypoxia. Brain natriuretic peptide (BNP) levels may also be elevated to varying degrees, correlating with disease severity and prognosis.
Electrocardiogram (ECG)
Although ECG cannot directly reflect elevated pulmonary arterial pressure, it can indicate right heart enlargement or hypertrophy.
Chest X-ray
Chest X-rays may show signs of pulmonary arterial hypertension:
- Enlargement of the right lower pulmonary artery trunk, with a transverse diameter ≥15 mm or a ratio of the right lower pulmonary artery diameter to the tracheal diameter ≥1.07. Dynamic observations may reveal a widening of the right lower pulmonary artery trunk by >2 mm.
- Prominent pulmonary artery segment or a height ≥3 mm.
- Central pulmonary artery dilation with peripheral pruning, forming a knuckle appearance.
- Significant convexity of the conus (right anterior oblique view at 45°) or a height ≥7 mm.
- Right ventricular enlargement.
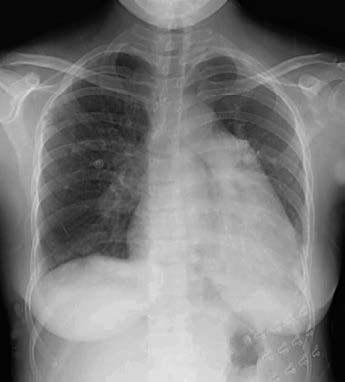
Figure 1 Frontal chest X-ray radiograph of pulmonary hypertension
Echocardiography and Doppler Ultrasound
Echocardiography is the most important non-invasive screening tool for pulmonary arterial hypertension. A tricuspid regurgitation peak velocity >3.4 m/s or pulmonary artery systolic pressure >50 mmHg suggests pulmonary arterial hypertension.
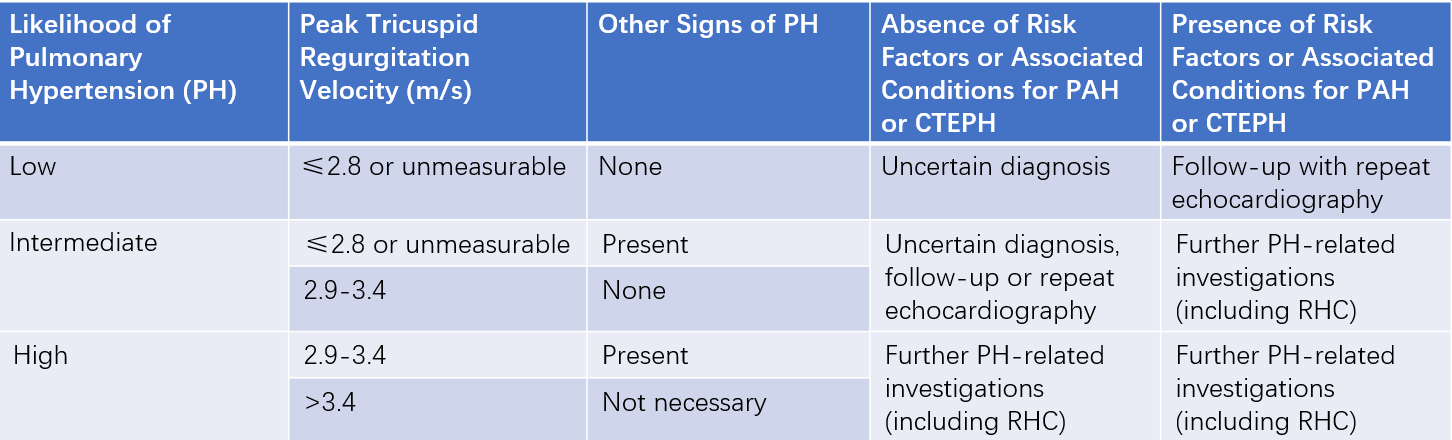
Table 1 Echocardiographic and Doppler ultrasound assessment of pulmonary hypertension (PH) and clinical recommendations
Note: Other echocardiographic signs of PH include findings related to the right ventricle, pulmonary artery, inferior vena cava, and right atrium.
Pulmonary Function Testing
Mild to moderate restrictive ventilatory defects and impaired diffusion capacity may be observed.
Arterial Blood Gas Analysis
Most patients exhibit mild to moderate hypoxemia due to ventilation-perfusion mismatch. Alveolar hyperventilation leads to reduced partial pressure of carbon dioxide (PaCO2). Severe hypoxemia may be associated with decreased cardiac output, pulmonary arterial thrombosis, or a patent foramen ovale.
Ventilation/Perfusion (V/Q) Scanning
IPAH patients may show diffuse perfusion defects or a normal scan. This is also a key method for excluding chronic thromboembolic pulmonary hypertension.
Right Heart Catheterization and Acute Vasoreactivity Testing
Right heart catheterization is considered the gold standard for diagnosing pulmonary arterial hypertension. It directly measures pulmonary arterial pressure, cardiac output, and pulmonary vascular resistance, and identifies left-to-right shunts, aiding in treatment planning.
Acute vasoreactivity testing evaluates the pulmonary vascular response to short-acting vasodilators and identifies patients who may benefit from oral calcium channel blockers. Agents used include inhaled iloprost, intravenous adenosine, and inhaled NO. A positive response is defined as a mean pulmonary arterial pressure (mPAP) reduction ≥10 mmHg to ≤40 mmHg, with unchanged or increased cardiac output. Generally, only 10-15% of IPAH patients meet this criterion.
Diagnosis and Differential Diagnosis
Pulmonary arterial hypertension (PAH) can be diagnosed when Doppler echocardiography estimates a pulmonary artery systolic pressure greater than 50 mmHg, combined with clinical findings. A definitive diagnosis of PAH requires right heart catheterization, with a mean pulmonary arterial pressure (mPAP) of at least 25 mmHg. IPAH is a diagnosis of exclusion, which can only be made after ruling out other causes of pulmonary arterial hypertension.
Treatment
The treatment strategy consists of the following components:
- Initial and supportive therapy.
- High-dose calcium channel blockers (CCBs) for patients with a positive acute vasoreactivity test, and targeted therapies for those with a negative test.
- Combination therapy or lung transplantation for patients with poor treatment response.
Initial Therapy
Women of childbearing age are advised to avoid pregnancy. Patients should receive timely influenza and pneumococcal vaccinations. Psychological and social support is provided. For patients with reduced physical capacity, rehabilitation is introduced alongside pharmacological treatment. If surgery is required, epidural anesthesia is preferred over general anesthesia.
Supportive Therapy
Oral Anticoagulants
Autopsy findings in IPAH patients indicate a high prevalence of in situ thrombus formation in the vasculature. Abnormalities in coagulation and fibrinolytic pathways have been reported. Non-specific risk factors for venous thromboembolism, such as heart failure and immobility, provide the theoretical basis for the use of oral anticoagulants.
Diuretics
Diuretics may be used to alleviate symptoms in cases of decompensated right heart failure with fluid retention, elevated central venous pressure, hepatic congestion, ascites, or peripheral edema.
Oxygen Therapy
Hypoxemia can induce pulmonary vasoconstriction, erythrocytosis, increased blood viscosity, and accelerated pulmonary arteriole remodeling, contributing to the progression of IPAH. Oxygen therapy is recommended for patients with WHO functional class III-IV symptoms or persistent arterial oxygen partial pressure (PaO2) below 8 kPa (60 mmHg) to maintain arterial oxygen saturation above 90%.
Digoxin
Digoxin improves cardiac output in IPAH and can be used to control ventricular rate in atrial tachyarrhythmias associated with PAH.
Anemia and Iron Status
Iron deficiency is associated with reduced exercise capacity and may correlate with higher mortality. Routine monitoring of iron status is recommended, and iron supplementation should be provided if deficiency is detected. Further investigation into the cause of iron deficiency is necessary.
Vasodilators
Calcium Channel Blockers (CCBs)
A positive acute vasoreactivity test is the indication for CCB therapy. CCBs are effective in only 10-15% of IPAH patients. Commonly used drugs include nifedipine, diltiazem, and amlodipine. Nifedipine is preferred for patients with bradycardia, while diltiazem is favored for those with tachycardia. Reassessment of treatment efficacy is performed in 3-4 months.
Prostacyclins
Prostacyclins not only dilate blood vessels and reduce pulmonary arterial pressure but also reverse vascular remodeling with long-term use. Common prostacyclin analogs include epoprostenol, iloprost, and beraprost. Additionally, prostacyclin receptor agonists such as selexipag are available.
Nitric Oxide (NO)
Inhaled NO selectively dilates pulmonary arteries without affecting the systemic circulation. However, its clinical application is limited due to its short duration of action and the inconvenience of preparing inhalable NO.
Endothelin Receptor Antagonists (ERAs)
Common ERAs include bosentan, ambrisentan, and macitentan.
Phosphodiesterase-5 (PDE-5) Inhibitors
Drugs in this category include sildenafil, tadalafil, and vardenafil.
Soluble Guanylate Cyclase (sGC) Stimulators
Riociguat is a commonly used sGC stimulator. The combined use of riociguat and PDE-5 inhibitors is not recommended.
Lung or Heart-Lung Transplantation
Lung transplantation is considered for patients who do not respond adequately to aggressive medical therapy. Patients with pulmonary veno-occlusive disease (PVOD) or pulmonary capillary hemangiomatosis (PCH) have a poor prognosis and lack effective medical treatments. A diagnosis of either condition warrants consideration of lung transplantation. Combined heart-lung transplantation may be appropriate in cases of irreversible structural or functional cardiac damage.
Health Guidance
Patients with IPAH benefit from lifestyle guidance and education to raise awareness of health-related knowledge. Confidence in managing the disease is encouraged. Preventing pulmonary infections is emphasized.