IgG4-related disease (IgG4-RD) is a chronic inflammatory autoimmune condition that can affect nearly all anatomical sites or organ systems, including glands, the orbit, sinuses, retroperitoneal space, pancreas, bile ducts, lungs, kidneys, lymph nodes, and even blood vessels and the pituitary gland. The peak incidence occurs between the ages of 50 and 60 years, with males accounting for 62.7%–65.3% of cases. The primary clinical manifestations include swelling or masses in the affected organs, along with elevated serum IgG4 levels and diffuse infiltration of IgG4+ plasma cells in the affected tissues. Typical pathological features include extensive plasma cell infiltration, storiform fibrosis, and obliterative phlebitis.
In 1995, Yoshida et al. introduced the concept of autoimmune pancreatitis (AIP) based on findings in patients with chronic pancreatitis. They noted that some patients exhibited diffuse pancreatic enlargement with histological evidence of lymphocyte infiltration and fibrosis, and responsiveness to steroid therapy. In 2000, Hamano discovered significantly elevated serum IgG4 levels in patients with AIP. It is now widely accepted that Type 1 AIP represents pancreatic involvement in IgG4-RD. Mikulicz disease, characterized by lacrimal and salivary gland inflammation, has also been confirmed to be an IgG4-related autoimmune disease.
Etiology and Pathogenesis
The etiology and mechanisms underlying IgG4-RD are believed to involve environmental factors, allergies, and infections.
Previous studies suggest that the activation of pattern recognition receptors (such as TLR or NLR) on antigen-presenting cells (APCs) due to pathogenic infection may act as a triggering factor for the development of IgG4-RD. Following receptor activation, cytokines such as BAFF and APRIL are secreted, which activate B cells and promote IgG4 production. The activated APCs also present antigens to CD4+ T cells, leading to their activation. Once activated, T cells differentiate into various subtypes, including Th1, Th2, Treg, and Tfh cells, which secrete cytokines such as IL-4, IL-10, IL-13, and IL-21. These cytokines further stimulate B cells to differentiate into plasmablasts and plasma cells, resulting in increased production of IgG4. Cytokines like IL-21 produced by Tfh cells can in turn amplify Tfh cell activation, facilitating the formation of ectopic germinal centers. This sequence of events leads to the infiltration of IgG4+ plasma cells in tissues and organs. Meanwhile, inflammatory cells producing large amounts of TGF-β and local fibroblast activation contribute to the progression of fibrosis.
Pathology
The hallmark pathological features include plasma cell infiltration, storiform fibrosis, and obliterative phlebitis. Plasma cells in the affected tissues exhibit high expression of IgG4, with the ratio of IgG4+ to IgG+ plasma cells typically exceeding 40%. Other findings may include lymphoid follicle and germinal center formation, eosinophilic infiltration, and varying degrees of lymphadenopathy, dacryoadenitis, and sialadenitis, which are associated with extensive IgG4+ plasma cell infiltration (commonly >50 cells/HPF, often >100 cells/HPF) and higher serum IgG4 levels. Peripheral blood eosinophilia may also accompany these manifestations.
In conditions like retroperitoneal fibrosis, sclerosing mesenteritis, and fibrosing mediastinitis, plasma cell infiltration in the tissues is relatively sparse, and germinal centers are less commonly observed. Storiform fibrosis tends to be more prominent in these scenarios.
Clinical Manifestations
The onset is often subacute, with clinical manifestations arising due to tissue or organ enlargement, compression, obstruction, or destruction of normal structures in the affected areas. The disease is capable of involving multiple systems throughout the body, resulting in diverse and variable symptoms of varying severity.
Pancreas
Type 1 autoimmune pancreatitis (AIP), also referred to as IgG4-related pancreatitis, accounts for 40%–60% of AIP cases. Pancreatic involvement is observed in 25%–36% of patients with IgG4-RD, with the primary clinical manifestation being diffuse pancreatic enlargement, often resembling a "sausage-like" appearance. Localized pancreatic masses may also occur, accompanied by pancreatic duct strictures, painless obstructive jaundice, or features mimicking pancreatic cancer. Pancreatic peripancreatic edema may result in the appearance of a "capsule-like rim." Patients typically do not experience acute pancreatitis episodes, but some may develop type 2 diabetes for the first time.
Lacrimal Glands and Orbits
Between 32% and 49% of patients present with swelling of the lacrimal glands and intraorbital soft tissue, accompanied by symptoms such as eyelid swelling, proptosis (eye protrusion), and diplopia (double vision), with vision impairment being relatively uncommon.
Salivary Glands and Other Glands
In 37% to 57% of cases, patients exhibit symmetrical enlargement of the submandibular and parotid glands, along with mild symptoms of dry mouth and dry eyes. Involvement of the salivary glands is often accompanied by lymphadenopathy in the submandibular and cervical regions.
Hepatobiliary System
Around 13% to 20% of patients experience involvement of the bile ducts, including extrahepatic, hilar, and/or intrahepatic biliary strictures, which manifest as obstructive jaundice or are associated with fever. Imaging may reveal biliary narrowing and stenosis, typically appearing as long-segment, smooth strictures causing proximal bile duct dilation. Biliary walls often show significant thickening and enhancement, with involvement primarily affecting the intrapancreatic segment of the common bile duct. The gallbladder may also exhibit thickening and enhancement. Hepatic involvement is rare, with an incidence of 2.2% reported in the literature, primarily manifesting as masses, often involving the hepatic hilum, and almost invariably associated with sclerosing cholangitis. Liver involvement is usually accompanied by abnormal liver function test results.
Urinary System
Presentations include flank pain, hydronephrosis, foamy urine, edema, and reduced urine output, potentially accompanied by renal insufficiency. Renal involvement occurs in 7.9%–15.6% of cases, most commonly as tubulointerstitial nephritis, which appears as diffuse renal enlargement with single or multiple low-density lesions. When the renal pelvis, ureters, or bladder are involved, associated findings may include soft tissue masses or focal thickening at the corresponding sites. Localized obstruction may lead to hydronephrosis. The prostate may also be affected.
Periaortitis/Retroperitoneal Fibrosis
This condition affects large arteries and their branches and is observed in 23%–26% of patients. It manifests as inflammation of vascular walls with infiltration of soft tissues around the vessels. The abdominal aorta below the renal arteries is most frequently involved, often extending to retroperitoneal tissues, kidneys, ureters, and other surrounding structures, causing retroperitoneal fibrosis. Typical symptoms include abdominal pain, lower back pain, edema, and hydronephrosis. The thoracic aorta, common iliac arteries, left and right iliac arteries, mesenteric arteries, common carotid arteries, subclavian arteries, and other parts of the aorta may also be affected, often resulting in aneurysmal dilation or aneurysm formation.
Lungs
Pulmonary involvement occurs in approximately 13%–29% of cases, with symptoms including dry cough, shortness of breath, and dyspnea. In most cases, patients are asymptomatic, with pulmonary abnormalities detected on imaging studies. These findings include inflammatory pseudotumors, tracheal/bronchial stenosis, interstitial pneumonia, and pleuritis.
Other
Lymph nodes, sinuses, mesentery, mediastinum, dura mater, pituitary gland, middle ear, larynx, and mastoid processes may show localized lesions or masses. Skin manifestations include erythema, papules, or maculopapular rashes. Allergic symptoms such as allergic rhinitis and asthma may also occur. Additionally, involvement of the bone marrow, heart, gastrointestinal tract, breasts, and gonads has been documented.
Laboratory Examinations
Elevated serum IgG4 levels are a characteristic finding. Some patients may also exhibit elevated IgE levels and peripheral eosinophilia.
Imaging exams, including ultrasound, CT, MRI, and PET-CT, assist with diagnosis, often revealing localized or diffuse tissue or organ enlargement, which may be difficult to distinguish from malignant tumors or inflammatory lesions. IgG4-related sclerosing cholangitis typically presents with irregular diffuse or localized bile duct or main pancreatic duct strictures. Parenchymal organ masses may show increased standardized uptake values (SUVs) on PET-CT. Histopathological examination is crucial for diagnosis and differentiation.
Diagnosis
The diagnostic criteria proposed by the Japan College of Rheumatology in 2012 and the ACR/EULAR classification criteria from 2019 are currently used.
2012 Diagnostic Criteria for IgG4-RD by the Japan College of Rheumatology
- Localized or diffuse enlargement of one or more organs detected by clinical examination.
- Elevated serum IgG4 levels (≥135 mg/dL).
- Histological findings:
- (a) Lymphocyte and plasma cell infiltration with fibrosis.
- (b) Infiltration of IgG4-positive plasma cells (IgG4+/IgG+ plasma cell ratio >40%, and IgG4+ plasma cells >10 per HPF).
Cases meeting all three criteria are definitive for IgG4-RD. Cases fulfilling criteria 1 and 3 are considered probable, and those meeting criteria 1 and 2 are considered possible.
2019 ACR/EULAR Diagnostic Criteria for IgG4-Related Disease
Inclusion Criteria: Characteristic clinical or radiological findings in typical organs (e.g., pancreas, salivary glands, bile ducts, orbit, kidney, lung, aorta, retroperitoneum, dura mater, or thyroid) where affected organs show enlargement or tumor-like masses. Exceptions include:
- Bile ducts, which more commonly present with strictures.
- Aorta, where typical findings include arterial wall thickening or aneurysmal dilation.
- Lungs, which often reveal thickened bronchovascular bundles.
Alternatively, pathologic evidence of plasma cell infiltration with accompanying inflammation of unknown cause in these organs may also meet the criteria.
Exclusion Criteria:
- Clinical: Fever, failure of glucocorticoid treatment.
- Serological: Unexplained lymphocytopenia or thrombocytopenia, peripheral eosinophilia, positive ANCA (especially PR3 or MPO antibodies), positive anti-SSA (Ro) or anti-SSB (La) antibodies, positive anti-double-stranded DNA, anti-RNP, or anti-Sm antibodies, presence of disease-specific autoantibodies, cryoglobulinemia.
- Radiological: Findings suggestive of tumor or infection, rapid progression on imaging, long bone pathologies consistent with Erdheim-Chester disease, splenomegaly.
- Pathological: Cellular infiltration suggesting malignancy, markers indicative of inflammatory myofibroblastic tumor, significant neutrophilic inflammation, necrotizing vasculitis, pronounced necrosis, granulomatous inflammation, or histological features consistent with macrophage/histiocyte-related diseases.
- Known Diagnoses: Diseases such as multicentric Castleman disease, Crohn’s disease or ulcerative colitis (if involvement is limited to the pancreaticobiliary system), Hashimoto’s thyroiditis (if thyroid involvement is isolated).
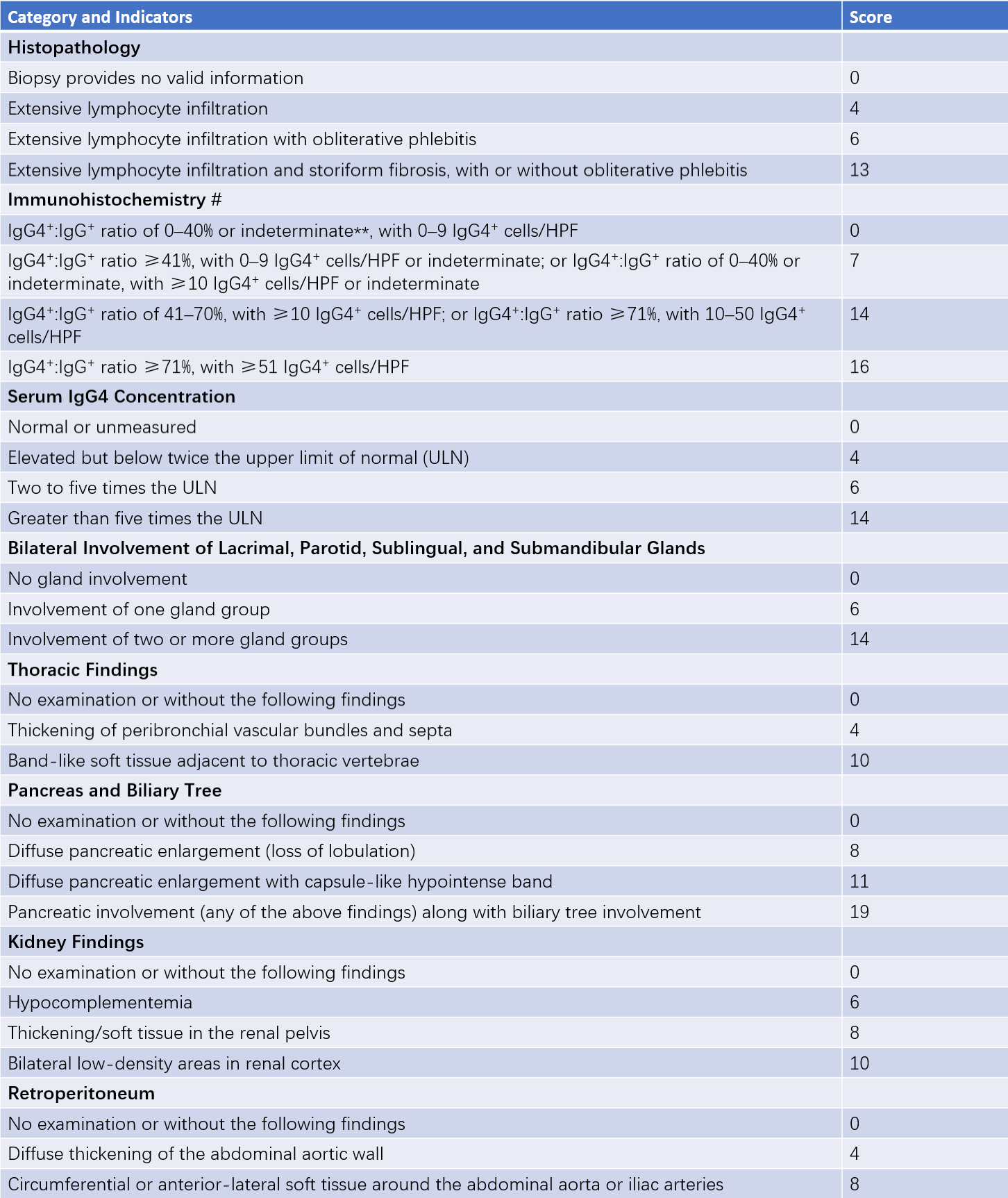
Table 1 2019 ACR/EULAR classification criteria for IgG4-RD
Notes:
Biopsy specimens from lymph nodes, gastrointestinal mucosa, and skin are not eligible for immunohistochemistry scoring.
"Indeterminate" refers to cases where the pathologist is unable to clearly quantify the number of positive-staining infiltrating cells but can confirm that the number is at least 10 cells per HPF. Due to various reasons, often related to the quality of immunohistochemical staining, the pathologist may be unable to precisely count the number of IgG4+ plasma cells. However, the pathologist can still reliably assign the case to the appropriate immunohistochemistry result group.
A diagnosis is made if the inclusion criteria are met, exclusion criteria are not satisfied, and the total score is ≥20.
Differential Diagnosis
Elevated IgG4 levels can be observed in various inflammatory diseases, neoplasms, and allergic disorders. Differential diagnoses include pancreatitis, lymphoma, plasmacytoma, Castleman disease, Rosai-Dorfman disease, histiocytic disorders, Crohn’s disease, primary sclerosing cholangitis, malignant tumors, infections, allergic diseases, and other autoimmune conditions. Histopathological examination is valuable for distinguishing IgG4-related disease from other conditions.
Treatment and Prognosis
Glucocorticoid therapy is effective for IgG4-related disease. Prednisone is typically administered at a dose of 30–40 mg per day [0.5–0.6 mg/(kg·d)], with tapering recommended after 2–4 weeks. The dose is reduced by 5 mg every 1–2 weeks until a maintenance dose is achieved, usually 2.5–5 mg per day, which is maintained for 1–3 years.
Immunosuppressive agents such as mycophenolate mofetil, leflunomide, cyclophosphamide [oral administration at 1–2 mg/(kg·d) or monthly intravenous infusion of 0.5–1.0 g/m2], azathioprine [1–2 mg/(kg·d)], iguratimod, and methotrexate may be added to the therapeutic regimen.
Treatment based on glucocorticoids is effective in 96% of patients. However, 33% experience recurrence during drug tapering, while the relapse rate after drug discontinuation is as high as 64%. CD20 monoclonal antibodies show efficacy in refractory or recurrent cases, reducing relapse rates but potentially increasing infection risks. Recent studies suggest that IL-6 receptor monoclonal antibodies also demonstrate promising therapeutic effects.
Most patients show favorable prognoses following appropriate treatment.