Antiphospholipid syndrome (APS) is an autoimmune disease primarily characterized by recurrent arterial and venous thrombosis, obstetric complications, and thrombocytopenia, accompanied by persistently positive antiphospholipid antibodies (APL) at moderate to high titers. The clinical manifestations of APS are complex and diverse, with involvement of multiple organ systems across the body. The most prominent feature is vascular thrombosis. APS is one of the most common causes of acquired thrombophilia, accounting for approximately 15%–20% of deep vein thrombosis cases, about one-third of new ischemic strokes in younger adults (aged <50 years), and 10%–15% of recurrent pregnancy loss cases. Additionally, 30%–40% of systemic lupus erythematosus (SLE) patients test positive for APL, and about 10%–15% exhibit APS-related clinical manifestations.
Etiology and Pathogenesis
The exact pathogenesis of APS remains incompletely understood. APL serves as the determinant factor in APS, with their primary target antigen being β2-glycoprotein I (β2GPⅠ). APL contribute to thrombus formation through multiple mechanisms:
- APL bind to phospholipids on vascular endothelial cell membranes, impairing endothelial cell function and reducing the synthesis and release of prostacyclin (PGI2).
- APL bind to platelet phospholipids, leading to thromboxane A2 release, platelet aggregation, and activation of platelet adhesion functions.
- APL directly interact with antithrombin III, protein C, protein S, and thrombomodulin, triggering coagulation via both intrinsic and extrinsic pathways while inhibiting the protein C system, ultimately promoting thrombus formation.
Clinical Manifestations
The clinical features of APS mainly fall into the following categories:
Thrombotic Events
The clinical manifestations of thrombosis in APS depend on the type, location, and size of the affected vessels, involving either single or multiple vessels. Venous thromboembolism is more common in APS, with the most frequent site being deep vein thrombosis of the lower limbs. Other involved sites may include the renal, hepatic, retinal veins, superior and inferior vena cava, and cerebral venous sinuses. Arterial thrombosis most commonly involves intracranial arteries but may also affect coronary, renal, and mesenteric arteries. Microvascular involvement includes conditions such as livedo reticularis, nephropathy, alveolar hemorrhage, and adrenal infarction.
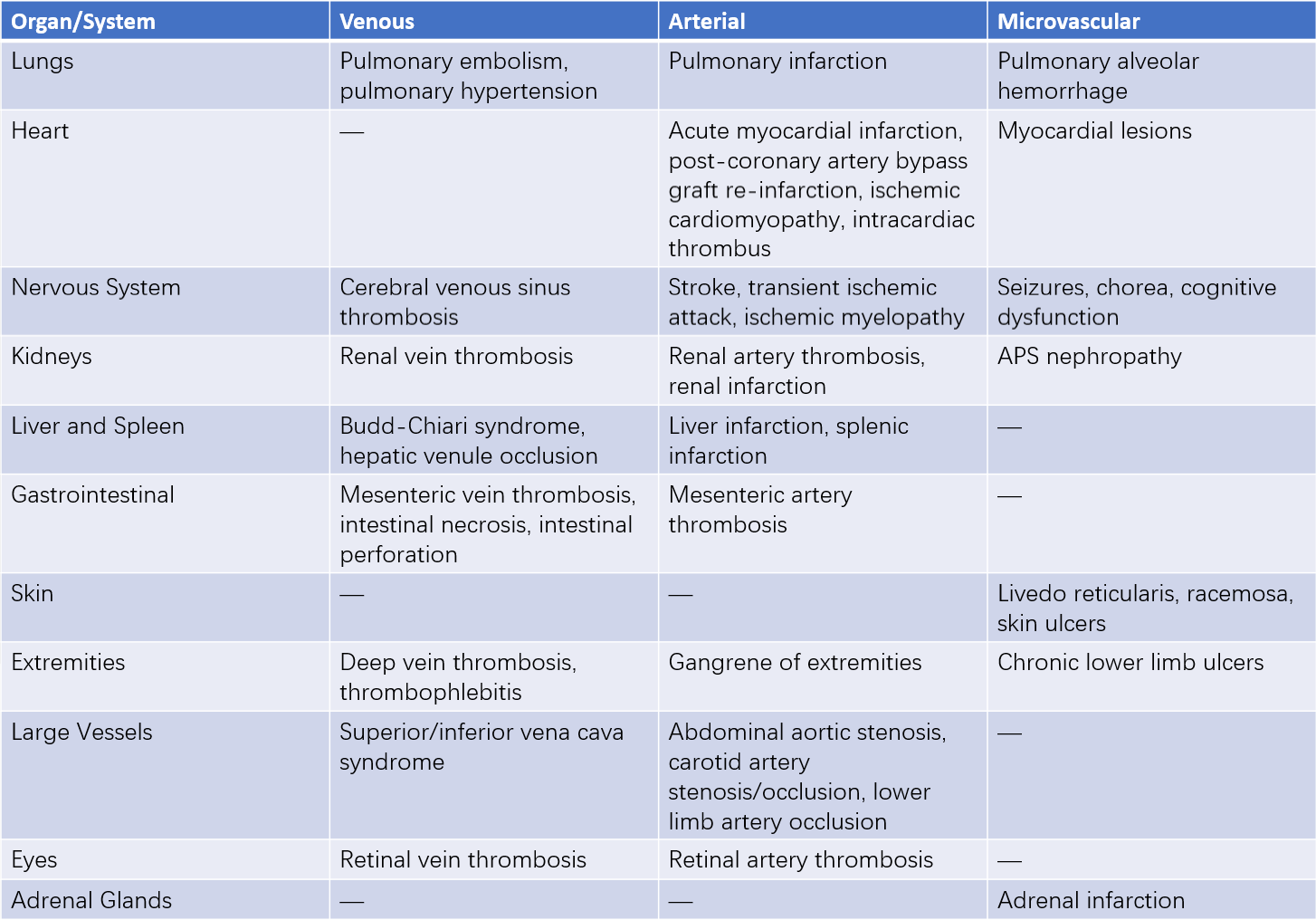
Table 1 Clinical manifestations of thrombosis in APS
A minority of APS patients may experience progressive thrombosis in three or more organs within one week, affecting vital organs such as the brain, kidneys, liver, or heart, leading to organ failure and death. This phenomenon, confirmed pathologically as thrombosis in small vessels, is referred to as catastrophic APS. Its incidence is approximately 1.0%, but the case fatality rate is as high as 50%–70%, often due to complications such as stroke, encephalopathy, hemorrhage, or infection. The suspected mechanisms include a "thrombotic storm" and a "cytokine storm."
Cardiac Valve Damage
Cardiac valve damage is one of the most common cardiac manifestations of APS. Clinical findings may include generalized valve thickening (>3 mm), localized thickening near the midsection of valve leaflets, irregular vegetations or nodules at the valve edges (Libman-Sacks endocarditis), or moderate to severe valve dysfunction (regurgitation, stenosis). The mitral valve is the most frequently affected, followed by the aortic valve. The differential diagnosis should exclude a history of rheumatic fever and infective endocarditis. Early-stage disease may present without significant symptoms or signs, and most cases are discovered during evaluation for severe valve damage or arterial thromboembolism. The pathogenesis may involve immune complex deposition on affected valves, leading to fibrin-platelet thrombi formation.
Hematologic Abnormalities
Hematologic manifestations include thrombocytopenia and hemolytic anemia. Thrombocytopenia is a common clinical feature of APS, with an incidence of 20%–53%, and is more frequent in APS associated with SLE. Thrombocytopenia is usually mild to moderate in severity, with possible mechanisms involving direct binding of APL to platelets, leading to activation and aggregation, thrombotic microangiopathy-related consumption, or excessive thrombus formation.
Obstetric Complications
Pregnancy loss in APS typically occurs after 10 weeks of gestation and is referred to as fetal demise. Early pregnancy loss may also occur, presenting as recurrent miscarriages, though these cases require differentiation from causes such as chromosomal abnormalities or endocrine/metabolic disorders. APS-related complications during mid-pregnancy include intrauterine growth restriction (IUGR), oligohydramnios, or stillbirth. Further progression can result in severe conditions such as preeclampsia or eclampsia, leading to preterm birth, or HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets).
Laboratory Tests
Routine examinations may reveal thrombocytopenia, autoimmune hemolytic anemia, reduced complement levels, mild proteinuria, or dysmorphic red blood cells. Specific laboratory markers include the following:
Antiphospholipid Antibody Panel
This refers to a group of autoantibodies targeting phospholipids and/or phospholipid-binding proteins. It is the most characteristic laboratory marker of APS and serves as a key predictor of both thrombotic events and obstetric complications in APS patients. Among these, lupus anticoagulant (LAC), anticardiolipin antibody (ACA), and anti-β2 glycoprotein I antibody (aβ2GPⅠ) are the laboratory criteria included in the classification of APS. Compared to ACA and aβ2GPⅠ, LAC exhibits stronger correlations with thrombosis and pregnancy morbidity. LAC testing is a functional assay that determines the presence of LAC based on its ability to prolong phospholipid-dependent coagulation times through different pathways in vitro.
Presence of Other Autoantibodies
Some patients may have overlapping autoimmune diseases, resulting in positive tests for antinuclear antibodies (ANA), anti-dsDNA antibodies, or anti-ENA antibodies.
Imaging Studies
Vascular color Doppler ultrasound can accurately detect arterial and venous thrombosis. Pulmonary ventilation/perfusion scintigraphy, spiral CT, and electron-beam CT pulmonary angiography can diagnose pulmonary thromboembolism. MRI can detect early microinfarctions in the brain.
Diagnosis
Diagnostic Criteria
APS should be suspected in the following scenarios:
- Unexplained thrombotic events.
- Recurrent thrombotic events.
- Thrombosis occurring in uncommon sites, such as mesenteric veins, hepatic veins, renal veins, or cerebral venous sinuses.
- Ischemic strokes or cardiovascular events in young individuals (<50 years old).
- Unexplained neurological symptoms, such as chorea, transverse myelopathy, or early-onset vascular dementia.
- Thrombosis in patients with SLE or other connective tissue diseases.
- Unexplained thrombocytopenia or autoimmune hemolytic anemia.
- Recurrent miscarriages or pregnancy complications associated with preterm delivery.
- Livedo reticularis or other thrombotic skin manifestations.
- Prolonged activated partial thromboplastin time (APTT) or false-positive results for syphilis serology detected incidentally during laboratory investigations.
APS is diagnosed based on a combination of clinical and laboratory criteria. According to the 2006 revised Sydney classification criteria for APS, a diagnosis requires at least one clinical criterion and one laboratory criterion.
Differential Diagnosis
The differential diagnosis of APS depends on its distinct clinical manifestations. Various acquired or hereditary factors can lead to pregnancy loss and/or thromboembolic diseases.
Venous thrombosis must be differentiated from inherited or acquired clotting disorders, such as protein C or protein S deficiency, factor V Leiden mutation, antithrombin deficiency, malignancies, myeloproliferative disorders, and nephrotic syndrome.
Arterial thrombosis requires differentiation from conditions such as atherosclerosis, embolic events, atrial fibrillation, atrial myxoma, infective endocarditis, fat embolism, thrombotic thrombocytopenic purpura, and systemic vasculitis.
Cases with simultaneous or sequential arterial and venous thrombosis should be assessed for heparin-induced thrombocytopenia, hypofibrinogenemia or fibrinogen-activating factor deficiencies, hyperhomocysteinemia, myeloproliferative disorders, polycythemia vera, paroxysmal nocturnal hemoglobinuria, Waldenström’s macroglobulinemia, sickle cell disease, or systemic vasculitis.
Treatment and Prognosis
The primary goals of treating APS are to prevent thrombosis and reduce the risk of pregnancy loss. Treatment should be personalized based on the patient's specific clinical manifestations, disease severity, and response to therapy. In addition to pharmacological treatment, management includes patient education to enhance adherence and lifestyle modifications such as smoking cessation and weight control.
Long-term and adequate anticoagulation is the cornerstone of therapy for thrombotic APS. Commonly used anticoagulants include vitamin K antagonists such as warfarin, as well as heparin or low-molecular-weight heparin. These may be used alone or in combination with antiplatelet agents like aspirin. Glucocorticoids and immunosuppressive agents are generally unnecessary for APS patients but may be used in cases of severe thrombocytopenia, hemolytic anemia, or microvascular involvement.
For obstetric APS, the choice of treatment depends on factors such as prior thrombotic events, history of pathological pregnancies, and coexisting connective tissue diseases (CTDs). Low-dose aspirin (75–100 mg per day) or a combination of low-dose aspirin and low-molecular-weight heparin may be selected.
For catastrophic APS, the first-line treatment includes anticoagulation with heparin combined with high-dose glucocorticoids, plasmapheresis, and/or intravenous immunoglobulin as a triple therapy. Concurrently, aggressive efforts are made to identify and manage potential triggers, including infectious sources or malignancies.
Overall, the prognosis for APS patients is relatively favorable, with a 10-year survival rate of approximately 90.7%. Major causes of mortality include thrombotic events, bleeding complications, and infection. Without proper treatment, thrombotic APS carries a recurrent thrombosis risk exceeding 50% within five years. For obstetric APS, untreated pregnancy success rates remain low at 10%–30%, but live birth rates can significantly improve to 70%–85% with appropriate treatment.