Congenital hearing loss refers to a type of hearing impairment that manifests at birth or shortly thereafter, caused by genetic factors or abnormalities during maternal gestation and delivery. It can be categorized into two major types: hereditary and non-hereditary. Based on pathological characteristics, it can also be classified as conductive, sensorineural, or mixed hearing loss.
Etiology and Clinical Manifestations
Hereditary Congenital Hearing Loss
Hereditary congenital hearing loss arises from genetic defects, such as gene mutations or chromosomal abnormalities, and is predominantly characterized as sensorineural hearing loss. It can be further divided into non-syndromic and syndromic types.
Non-Syndromic Type
This type only involves hearing loss, accounting for 70% of hereditary hearing loss cases.
Syndromic Type
In addition to hearing loss, this type is accompanied by malformations of the external ear or abnormalities in other organs and systems, including the heart, kidneys, nervous system, visual system, maxillofacial and skeletal systems, endocrine system, or skin. Syndromic hearing loss accounts for 30% of hereditary hearing loss. Examples include Usher syndrome (associated with retinitis pigmentosa), Treacher-Collins syndrome (characterized by mandibulofacial dysostosis with skeletal defects), Crouzon disease (craniofacial dysostosis), and Waardenburg syndrome (featuring sensorineural hearing loss along with pigmentation abnormalities in other tissues and organs).
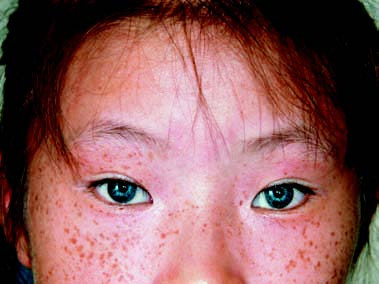
Figure 1 Facial features of a patient with Waardenburg syndrome
Currently, genetic mutations linked to hereditary hearing loss have been identified across various gene families responsible for diverse functions, such as transcription factors, cytoskeletal elements, and ion channels, reflecting the genetic heterogeneity and complexity of the condition.
Autosomal dominant hereditary hearing loss accounts for approximately 15–20% of non-syndromic cases, often presenting as postlingual sensorineural hearing loss. However, the age of onset varies based on the specific gene mutation, ranging from congenital to adulthood. Examples include dominant mutations such as DFNA3, DFNA8, DFNA12, DFNA6, DFNA13, DFNA14, and DFNA38.
Autosomal recessive inheritance accounts for 70–80% of non-syndromic hearing loss cases, typically manifesting as congenital sensorineural hearing loss, but variations in age of onset may occur depending on the gene mutation. For instance, DFNB8, an autosomal recessive type, predominantly causes postlingual sensorineural hearing loss.
Sex-linked and mitochondrial inheritance are relatively rare, accounting for less than 1% of hereditary hearing loss.
Common genetic mutations involved in hearing loss that are widely tested in clinical practice include GJB2 (DFNB1 and DFNA3), SLC26A4 (DFNB4), GJB3, and mitochondrial 12S rRNA mutations (e.g., m.A1555G or m.C1494T).
GJB2 gene follows an autosomal recessive inheritance pattern and is linked to approximately 20% of childhood hearing loss. Mutations often cause severe or profound congenital hearing loss. Patients with mutations in this gene typically respond well to hearing rehabilitation with cochlear implants.
SLC26A4 gene is also autosomal recessive and accounts for about 15% of childhood hearing loss cases. It is associated with congenital inner ear malformations such as enlarged vestibular aqueduct (EVA) syndrome or Mondini dysplasia. Patients may have partial hearing loss at birth or initially normal hearing that progressively deteriorates. Hearing decline may also occur in response to triggers such as colds, coughing, sneezing, head trauma, or physical impact, potentially leading to severe to profound hearing loss or total deafness over time.
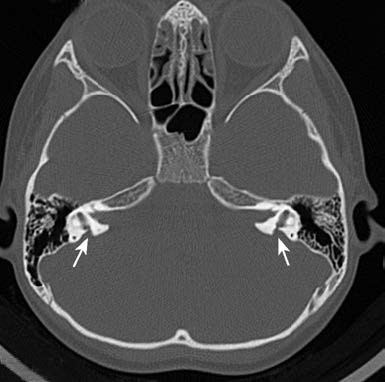
Figure 2 Temporal bone CT of a patient with enlarged vestibular aqueduct syndrome
GJB3 gene may follow either autosomal recessive or dominant inheritance patterns and is often associated with acquired high-frequency hearing loss. Researchers successfully cloned the GJB3 gene in 1998.
Pathogenic variants of mitochondrial MT-RNR1 mutations are strongly associated with susceptibility to aminoglycoside antibiotics. Specific hotspot mutations, such as m.A1555G and m.C1494T, occur in 2–11% of individuals with hearing loss in China. Even when exposed to standard or minimal doses of aminoglycosides, individuals carrying these mutations may experience permanent hearing loss.
Genetic screening for hearing loss plays a significant role in the prevention and treatment of hereditary congenital hearing loss. It provides insight into the causes of hearing loss for some patients, clarifies the carrier status of pathogenic genes within affected families, and offers valuable information for clinical genetic counseling, prenatal diagnosis, and preventive strategies. These screenings contribute to reducing the birth of infants with hearing loss and improving early diagnosis and intervention outcomes.
Nonhereditary Congenital Hearing Loss
Nonhereditary congenital hearing loss refers to hearing impairment caused by pathological factors affecting the child during the embryonic development period, perinatal period, or delivery. Such factors may include maternal infections, poisoning, hypoxia, or trauma. The condition is typically associated with the following causes:
Medication Factors
Improper use of certain medications by the mother during pregnancy (e.g., aminoglycosides, cytotoxic drugs, antimalarials, and diuretics) may allow these drugs to cross the placenta and enter the fetus, leading to fetal toxicity and hearing loss.
Disease-Related Damage
During the prenatal period, maternal infections with specific pathogens (e.g., gonorrhea, syphilis, HIV) may damage the fetal auditory system via placental transmission. This can cause injury to the cochlea, vestibule, or auditory nerve, or lead to viral or bacterial labyrinthitis, resulting in inner ear malformations or abnormalities and subsequent hearing loss.
During the postnatal period, conditions such as prematurity, severe asphyxia during birth, low birth weight, high bilirubin levels (hyperbilirubinemia), or septic meningitis may impair the auditory system in newborns, contributing to hearing loss.
Birth Trauma
During childbirth, improper use of forceps may result in trauma to the fetal skull, causing damage to the auditory organs.
Diagnosis
A comprehensive diagnosis involves systematically collecting the patient’s medical history, personal history, and family history. This process is supplemented by thorough clinical physical examinations and audiological assessments. Laboratory investigations, including imaging studies, hematological tests, immunological evaluations, and genetic testing, may provide scientific evidence for identifying the causes and types of congenital hearing loss.
Prevention
Prevention plays a more important and effective role compared to treatment.
Genetic Counseling and Prenatal Diagnosis
Promoting genetic counseling and advocating for better-quality reproductive health measures are crucial steps. Utilizing modern scientific technologies such as genetic testing improves hearing loss screening and diagnostics, enabling prenatal diagnosis of hereditary hearing loss and helping to reduce its incidence rates.
Maternal and Child Healthcare
Strengthening maternal and child healthcare during pregnancy and perinatal periods can prevent pregnancy-related diseases and reduce the likelihood of birth injuries. Conducting hearing screenings for infants after birth facilitates the early identification and management of hearing impairments.
Preventing Infectious and Nutritional Deficiency Diseases
Efforts to prevent infectious diseases and nutritional deficiencies, along with minimizing exposure to harmful physical factors such as excessive noise and toxic chemicals, contribute to health preservation. Lifestyle adjustments, including avoiding smoking and alcohol consumption and maintaining physical fitness, enhance resistance to factors that cause hearing loss.
Avoiding Ototoxic Medications
Medications that may damage hearing should be used with precise indications and under strict control regarding dosage and duration. During medication use, monitoring hearing status becomes essential, and any signs of hearing damage should prompt immediate discontinuation and appropriate treatment.
Treatment
The general approach focuses on early detection and diagnosis, followed by timely intervention. Restoring or partially restoring the lost hearing ability is prioritized, as well as preserving and utilizing residual hearing. Conductive hearing loss caused by congenital abnormalities of the external or middle ear can often be managed through surgical correction or the use of hearing aids. However, no clinically approved drug treatments are currently available for congenital sensorineural hearing loss. For individuals with residual hearing, early fitting of suitable hearing aids is recommended, and implantable hearing devices or cochlear implants can be explored in eligible cases. Hearing and speech rehabilitation is emphasized to reduce the risk of further complications such as deaf-mutism. Specific treatments are outlined as follows:
Medications
Timely and appropriate medication use in the early stages is critical for successful outcomes. Drug selection is based on the underlying cause and type of hearing loss. For hereditary hearing loss with identified genetic defects at the molecular level, gene therapy approaches may be explored. For hearing loss caused by viral or bacterial infections, early intervention with antiviral or antibacterial drugs may be attempted. Common supportive treatments for hearing rehabilitation include vasodilators, anticoagulants, thrombolytics, neurotrophic agents, and energy-boosting preparations, which may be used selectively based on clinical findings.
Hearing Aids and Middle Ear Implants
Hearing aids and middle ear implants are devices designed to amplify sound intensity, serving as key tools for auditory intervention in congenital hearing loss patients. Proper selection of these devices requires detailed examinations by otologists or audiologists. Hearing aids typically consist of a miniature microphone, amplifier, earphones, earmolds, and a power supply. Middle ear implants include components such as a microphone, amplifier, speech processor, signal transmission systems, and output sensors.
Cochlear Implantation
As a major intervention for congenital sensorineural hearing loss, cochlear implantation is primarily suited for individuals who do not benefit from high-power hearing aids, show no active ear disease, and have no severe inner ear malformations or auditory nerve absence based on imaging evaluations. The device comprises two main parts: an external unit (directional microphone, speech signal processor, and transmitter) and an internal unit (receiver, decoder, and stimulation electrodes).
Auditory and Speech Training
Auditory Training
This involves utilizing residual hearing or reconstructed hearing through hearing aids or cochlear implants. By providing long-term, structured auditory stimuli, the training aims to develop auditory habits and improve abilities such as auditory awareness, attention, localization, recognition, and memory.
Speech Training
Using complementary sensory functions such as hearing, vision, and touch, speech training employs scientific methods and appropriate devices (e.g., audiometers, speech analyzers) to improve speech production, lip-reading, vocabulary acquisition, grammatical understanding, and communication skills. With the widespread use of cochlear implants, more emphasis is placed on integrating auditory aids into speech training for better outcomes.
Surgical Interventions
Certain types of conductive hearing loss caused by middle ear abnormalities may be addressed through hearing reconstruction surgeries based on the cause, location, nature, and extent of the condition.
Gene Therapy
Gene therapy, an emerging biological treatment approach, targets specific genetic defects for compensation or correction and holds significant promise for curing hereditary hearing loss. For instance, mutations in the OTOF gene often result in prelingual, severe-to-profound, or total deafness in infants and children. Advances in gene-editing technologies, such as CRISPR-Cas9, have introduced new hope for treatment by paving the way for potential gene therapy applications for hearing loss.