Acute ST-segment elevation myocardial infarction (STEMI) is acute myocardial ischemic necrosis, primarily caused by a sudden and severe reduction or interruption of coronary blood supply, leading to profound and sustained myocardial ischemia. The most common cause is persistent and complete coronary artery occlusion resulting from thrombus formation secondary to rupture, erosion, or ulceration of unstable coronary plaques. Less commonly, it may result from coronary ostial obstruction, such as in cases of aortic dissection involving the coronary ostium or iatrogenic causes like coronary ostial obstruction during transcatheter aortic valve implantation (TAVI). Other rare causes include thromboembolism, spontaneous coronary artery dissection (SCAD), and prolonged coronary spasm.
Etiology and Pathogenesis
The fundamental cause of STEMI is acute occlusion of one or more coronary arteries on the basis of atherosclerosis. If the occlusion persists for 20-30 minutes or longer, it can result in AMI (Type 1 myocardial infarction). Extensive research has shown that most cases of STEMI are due to the rupture of unstable atherosclerotic plaques, followed by intraplaque hemorrhage and thrombus formation, leading to vascular occlusion.
Factors that precipitate plaque rupture, hemorrhage, and thrombus formation include:
- Increased sympathetic activity between 6 a.m. and 12 p.m., which heightens stress responses, increases myocardial contractility, heart rate, blood pressure, and coronary tension
- Postprandial hyperlipidemia, particularly after consuming large amounts of fat, which raises blood lipid levels and viscosity
- Intense physical exertion, emotional stress, or straining during defecation, all of which significantly increase left ventricular load
- Conditions such as shock, dehydration, hemorrhage, surgery, and severe arrhythmias, which lead to a sudden drop in cardiac output and a sharp reduction in coronary perfusion
STEMI can occur in patients with frequent angina or in those without prior symptoms. Severe complications following STEMI, such as arrhythmias, shock, and heart failure, can further reduce coronary perfusion, enlarging the area of myocardial necrosis.
Recent studies indicate that approximately 14% of STEMI patients undergoing coronary angiography exhibit no significant obstruction, a condition termed MINOCA (myocardial infarction with non-obstructive coronary arteries). Causes include plaque rupture or erosion, coronary spasm, coronary thromboembolism (potentially self-resolving), SCAD, Takotsubo cardiomyopathy (stress cardiomyopathy), and other forms of Type 2 AMI (e.g., anemia, tachy-brady syndrome, respiratory failure, hypotension, shock, severe hypertension with or without left ventricular hypertrophy, severe aortic valve disease, heart failure, cardiomyopathy, or drug/toxin-induced injury). Treatment strategies for these patients differ from those with obstructive coronary artery disease and require early identification and individualized treatment based on the underlying cause.
Pathology
The majority of STEMI patients exhibit thrombus formation superimposed on atherosclerotic plaques, leading to vascular occlusion. However, in rare cases of coronary occlusion caused by spasm, significant atherosclerotic lesions may not be present. The occurrence of infarction does not necessarily correlate with the number of coronary vessels affected by atherosclerosis or the degree of luminal stenosis.
Left anterior descending artery (LAD) occlusion leads to infarction of the anterior wall, apex, anteroseptal wall, and anterior papillary muscle of the left ventricle.
Right coronary artery (RCA) occlusion causes infarction of the inferior wall of the left ventricle (in RCA-dominant hearts), posterior interventricular septum, and right ventricle. It may also involve the sinoatrial (SA) node and atrioventricular (AV) node.
Left circumflex artery (LCX) occlusion results in infarction of the high lateral wall, inferior wall (in LCX-dominant hearts), and left atrium, potentially involving the AV node.
Left main coronary artery (LMCA) occlusion causes extensive infarction of the left ventricle.
Infarction of the right ventricle and left/right atrium is less common.
Within 20-30 minutes of coronary occlusion, a small portion of the myocardium supplied by the affected artery undergoes necrosis, initiating the pathological process of AMI. Between 1-2 hours, the majority of the myocardium in the affected region undergoes coagulative necrosis, accompanied by congestion, edema, and significant infiltration of inflammatory cells in the myocardial interstitium. Necrotic myocardial fibers gradually dissolve, forming zones of myocytolysis, followed by the development of granulation tissue.
Under intracavitary pressure, the necrotic myocardial wall may bulge outward, leading to cardiac rupture (e.g., free wall rupture, ventricular septal perforation, or papillary muscle rupture) or the gradual formation of a ventricular aneurysm. Necrotic tissue begins to be resorbed in 1-2 weeks and gradually undergoes fibrosis, forming a scar within 6-8 weeks, known as old myocardial infarction.
Pathophysiology
Acute myocardial infarction primarily leads to left ventricular diastolic and systolic dysfunction, resulting in various hemodynamic changes. The severity and duration of these changes depend on the location, extent, and size of the infarction. Key pathophysiological alterations include weakened myocardial contractility, reduced compliance, and uncoordinated myocardial contraction. These changes lead to a decrease in the maximum rate of pressure rise in the left ventricle (dp/dt), an increase in left ventricular end-diastolic pressure, and an increase in both end-diastolic and end-systolic volumes. Ejection fraction decreases, stroke volume and cardiac output decline, and heart rate increases, which are often accompanied by arrhythmias and blood pressure reduction. In severe cases, arterial oxygen content decreases.
In patients with extensive AMI, pump failure may occur, leading to cardiogenic shock or acute pulmonary edema. Right ventricular AMI is less common and is characterized by acute right heart failure with hemodynamic abnormalities, including elevated right atrial pressure exceeding left ventricular end-diastolic pressure, reduced cardiac output, and hypotension.
Ventricular remodeling, a subsequent change following AMI, involves left ventricular enlargement, shape alteration, thinning of the infarcted myocardial segment, and compensatory thickening of the non-infarcted myocardium. These changes have persistent effects on ventricular contractility and electrical activity, necessitating prompt clinical intervention to prevent adverse remodeling.
Clinical Manifestations
The clinical presentation of STEMI is closely related to the size, location of the infarction, and the status of coronary collateral circulation.
Prodromal Symptoms
50% - 81.2% of patients experience prodromal symptoms days before the onset of AMI, such as fatigue, chest discomfort, palpitations, dyspnea, restlessness, and angina during exertion. Among these, new-onset angina or worsening angina are the most prominent. Timely hospitalization and management during this phase can prevent AMI in some patients.
Symptoms
Pain is the most common initial symptom, often occurring in the early morning. Its location and characteristics are similar to angina pectoris, but it typically lacks an obvious trigger, occurs at rest, is more severe, and lasts longer—often for several hours or more. Rest and sublingual nitroglycerin usually fail to relieve the pain. Patients often exhibit restlessness, perspiration, fear, chest tightness, or a sense of impending doom.
Some patients may initially present with shock or acute heart failure without experiencing pain. Pain may also localize to the upper abdomen, leading to misdiagnosis as gastrointestinal emergencies such as perforated peptic ulcer and acute pancreatitis. Pain radiating to the jaw, neck, or upper back may be mistaken for dental or musculoskeletal issues.
Systemic symptoms such as fever, tachycardia, leukocytosis, and elevated erythrocyte sedimentation rate (ESR) occur due to the absorption of necrotic material. These symptoms typically appear 24-48 hours after the onset of pain and correlate with the extent of infarction. Fever generally reaches around 38°C and rarely exceeds 39°C, lasting approximately one week.
Severe pain is often accompanied by frequent nausea, emesis, and upper abdominal distension, which are related to vagus nerve stimulation by necrotic myocardium and reduced cardiac output leading to tissue hypoperfusion. Intestinal bloating is also common, and severe cases may present with hiccups.
Arrhythmias occur in 75%-95% of patients, most commonly within the first 1-2 days, with the highest incidence in the first 24 hours. Symptoms may include fatigue, dizziness, and syncope.
Ventricular arrhythmias are the most common, including frequent premature ventricular contractions (more than 5 per minute), paired PVCs, short bursts of ventricular tachycardia, and multifocal PVCs. PVCs occurring during the vulnerable period of the preceding beat (R-on-T phenomenon) are often precursors to ventricular fibrillation. Ventricular fibrillation is a leading cause of early death in STEMI, particularly before hospital admission. Atrioventricular (AV) block and bundle branch block are also common, while supraventricular arrhythmias are less frequent and typically occur in patients with heart failure. Anterior wall AMI with AV block indicates extensive infarction and severe disease.
Blood pressure often drops during pain but does not necessarily indicate shock. Persistent hypotension (systolic blood pressure <80 mmHg) after pain relief, accompanied by symptoms such as restlessness, pallor, cold and clammy skin, rapid and weak pulse, diaphoresis, reduced urine output (<20 mL/h), lethargy, or syncope, indicates shock.
Shock typically occurs within several hours to days after onset and affects approximately 20% of patients. The primary cause is cardiogenic shock, resulting from extensive myocardial necrosis (>40%) and a dramatic reduction in cardiac output. Peripheral vasodilation due to neurogenic reflexes is a secondary factor. In some cases, hypovolemia may also contribute.
Heart failure in the context of AMI is primarily acute left heart failure, which can occur within the first few days after onset or during the recovery phase from pain or shock. It is caused by significant weakening or uncoordinated contraction of the heart following AMI, with an incidence of approximately 32%-48%. Symptoms include dyspnea, cough, cyanosis, and restlessness. Severe cases may develop pulmonary edema, and subsequently, signs of right heart failure such as jugular vein distension, hepatomegaly, and peripheral edema may appear. In patients with right ventricular AMI, signs of right heart failure (e.g., elevated jugular venous pressure) may occur early, and can be often accompanied by hypotension.
Based on the presence and severity of heart failure symptoms and corresponding hemodynamic changes, heart failure caused by AMI can be classified using the Killip classification:
- Killip Class I: No signs of heart failure
- Killip Class II: Left heart failure with cracklings in less than 50% of the lung fields
- Killip Class III: Acute pulmonary edema with diffuse cracklings (wet and dry) throughout the lung fields
- Killip Class IV: Cardiogenic shock with various degrees or stages of hemodynamic compromise
In STEMI, severe left ventricular failure or pulmonary edema and cardiogenic shock are both caused by left ventricular systolic dysfunction. These two conditions often coexist to varying degrees and are collectively referred to as pump failure.
From a hemodynamic perspective, pulmonary edema is primarily associated with elevated left ventricular end-diastolic pressure, left atrial pressure, and pulmonary capillary wedge pressure (PCWP). Shock, on the other hand, is characterized by reduced cardiac output and arterial pressure. Cardiogenic shock represents a more severe form of pump failure than left ventricular failure, with a significantly lower cardiac index (CI) despite adequate left ventricular filling.
Forrester et al. revised these hemodynamic classifications and correlated them with clinical presentations, dividing patients into four categories:
- Class I: No pulmonary congestion or peripheral hypoperfusion; PCWP and CI are normal.
- Class II: Pulmonary congestion only; PCWP is elevated (≥18 mmHg), but CI remains normal (>2.2 L/min/m2).
- Class III: Peripheral hypoperfusion only; PCWP is normal (<18 mmHg), but CI is reduced (≤2.2 L/min/m2), often associated with hypovolemia or bradycardia.
- Class IV: Both pulmonary congestion and peripheral hypoperfusion; PCWP is elevated (≥18 mmHg), and CI is reduced (≤2.2 L/min/m2).
Among these classifications, Class IV is the most severe in both systems.
Signs
The cardiac dullness area may be normal or mildly to moderately enlarged. Tachycardia is common, though bradycardia may occur in some cases. The first heart sound (S1) at the apex is often diminished, and a fourth heart sound (atrial gallop) is frequently heard. A third heart sound (ventricular gallop) may also occur in some cases, indicating heart failure. Pericardial friction rub may develop in 10%-20% of patients on the 2nd or 3rd day after onset, which is caused by reactive fibrinous pericarditis. A coarse systolic murmur or a mid-to-late systolic click at the apex may indicate papillary muscle dysfunction or rupture. A new coarse systolic murmur with a palpable thrill at the left sternal border (3rd-4th intercostal space) suggests ventricular septal rupture.
Various arrhythmias may also be observed. Close monitoring of physical signs during the course of AMI is essential to promptly detect and identify mechanical complications.
Apart from a transient increase in blood pressure during the very early stage, almost all patients experience a drop in blood pressure. In patients with pre-existing hypertension, blood pressure may return to normal levels and may not recover to pre-AMI levels, indicating impaired cardiac pump function.
Additional signs related to arrhythmias, shock, or heart failure may also be present.
Laboratory and Auxiliary Examinations
Electrocardiogram (ECG)
ECG often shows progressive dynamic changes and is highly valuable for diagnosing AMI, determining its location and extent, assessing disease progression, and estimating prognosis. For patients with acute chest pain, performing an ECG within 10 minutes of first medical contact (FMC) is crucial to promptly confirm the diagnosis of STEMI.
Characteristic changes include
- ST-segment elevation: Convex upward elevation appears in leads facing the myocardial injury zone surrounding the necrotic area
- Wide and deep Q waves (pathological Q waves): These appear in leads facing the transmural myocardial necrosis zone
- T-wave inversion: It occurs in leads facing the ischemic zone surrounding the injury area
Leads opposite the AMI zone show reciprocal changes (mirror-image changes), such as R-wave elevation, ST-segment depression, and upright, tall T waves.
Dynamic Changes
During the first few hours, the ECG may appear normal or show abnormally tall, asymmetrical T waves, indicating the hyperacute phase.
A few hours later, the ST segment becomes significantly convex upward, merging with the upright T wave to form a monophasic curve. Within several hours to 2 days, pathological Q waves appear, and R waves diminish, marking the acute phase. Pathological Q waves stabilize within 3-4 days and persist permanently in 70%-80% of patients.
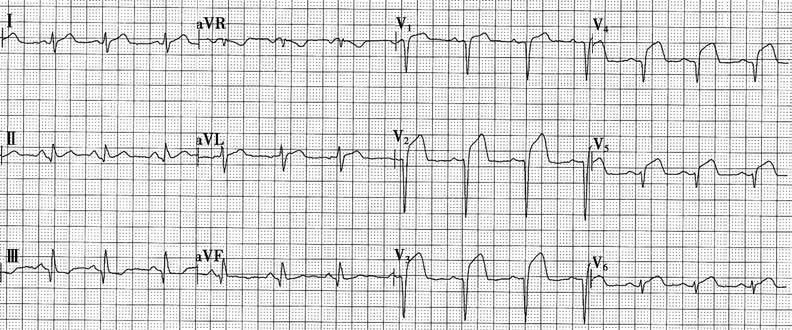
Figure 1 Electrocardiogram of acute anterior wall myocardial infarction
The figure shows QRS complexes in leads V1-V5 presenting as QS patterns, with significant ST-segment elevation.
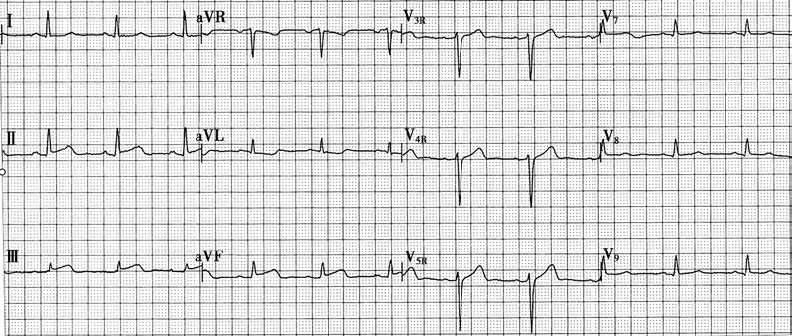
Figure 2 Electrocardiogram of acute inferior wall myocardial infarction
The figure shows ST-segment elevation in leads II, III, and aVF.
Without intervention, ST-segment elevation persists for several days to about two weeks before gradually returning to baseline, while T waves become flattened or inverted, indicating the subacute phase. Persistent ST-segment elevation suggests the formation of a ventricular aneurysm.
Weeks to months later, T waves become symmetrically inverted with a sharp, pointed nadir, indicating the chronic phase. T-wave inversion may persist permanently or gradually recover over months to years.
The location and extent of STEMI can be determined based on the number and pattern of leads showing characteristic changes.
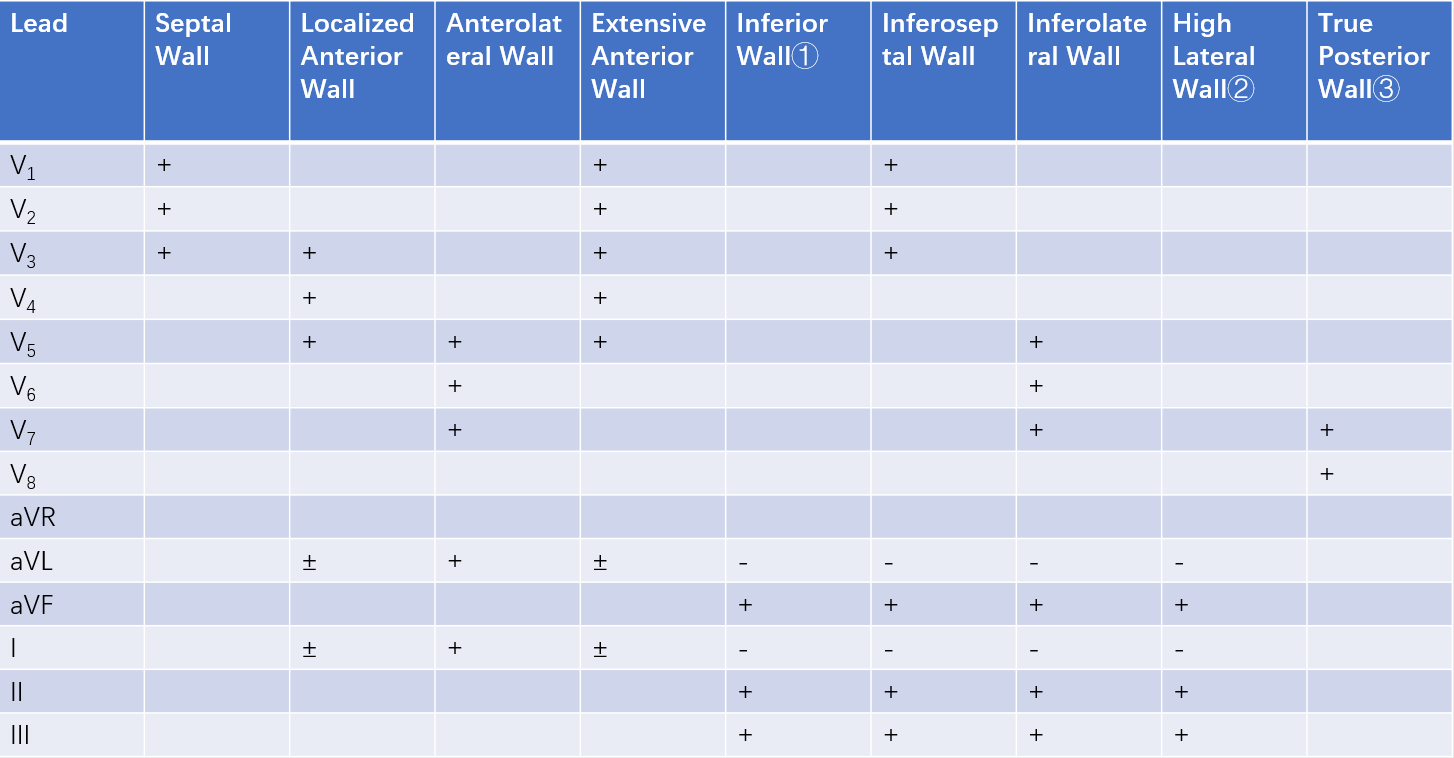
Table 1 Electrocardiographic localization of ST-elevation myocardial infarction (STEMI)
① Refers to the diaphragmatic surface. Right ventricular myocardial infarction (MI) is difficult to diagnose on a standard ECG, but ST-segment elevation in lead CR4R (negative electrode on the right upper arm, positive electrode at the V4 position) or V4R can serve as a reference for inferior wall MI extending to the right ventricle.
② Changes may be observed in leads V5, V6, and V7 located 1-2 intercostal spaces.
③ Tall R waves may appear in leads V1, V2, and V3. Similarly, tall R waves may appear in leads V1 and V2 during anterolateral infarction.
“+”: Positive changes, indicating typical ST-segment elevation, Q-wave, and T-wave alterations.
“-”: Reciprocal changes, indicating upward deflection of the main QRS wave, ST-segment depression, and T-wave inversion opposite to the “+” leads.
“±”: Possible positive changes.
Echocardiography
Two-dimensional and M-mode echocardiography can assess ventricular wall motion and left ventricular function. It is useful for diagnosing complications such as ventricular aneurysm, papillary muscle dysfunction, pericardial effusion, and ventricular septal rupture.
Cardiac Magnetic Resonance Imaging (CMR)
CMR can evaluate myocardial edema, necrosis, and perfusion abnormalities in AMI, and myocardial fibrosis scars in old MI. It also assesses ventricular size, systolic function, and regional wall motion abnormalities. CMR is helpful in diagnosing AMI and detecting complications such as pericardial effusion, cardiac rupture, and ventricular septal rupture.
Radionuclide Imaging
Positron emission tomography (PET) can observe myocardial metabolic changes and is currently the only imaging technique capable of directly assessing myocardial viability.
Single photon emission computed tomography (SPECT), combined with ECG-gated blood pool imaging, can evaluate wall motion, wall thickness, and overall cardiac function.
Laboratory Tests
Complete Blood Count (CBC)
Within 24-48 hours after onset, white blood cell counts may increase to (10-20) × 109/L, with neutrophilia and a decrease or disappearance of eosinophils. Erythrocyte sedimentation rate (ESR) increases, and C-reactive protein (CRP) levels rise, persisting for 1-3 weeks. Free fatty acid levels in the blood increase within hours to 2 days after onset.
Serum Cardiac Biomarkers
Elevated levels of cardiac biomarkers are strongly correlated with the extent of myocardial necrosis and prognosis:
- Myoglobin rises within 2 hours after onset, peaks at 12 hours, and returns to normal within 24-48 hours
- cTnI rises 3-4 hours after onset, peaks at 11-24 hours, and normalizes within 7-10 days
- cTnT peaks at 24-48 hours and normalizes within 10-14 days
- Creatine kinase-MB (CK-MB) rises within 4 hours after onset, peaks at 16-24 hours, and normalizes within 3-4 days
Elevated troponin levels are highly sensitive indicators for diagnosing MI.
CK-MB levels accurately reflect the extent of infarction, and an earlier peak suggests successful thrombolytic therapy.
Myoglobin rises earliest after AMI and is highly sensitive but lacks specificity. Troponins are highly specific but rise slightly later. If troponin levels are negative within 6 hours after symptom onset, retesting in 1-2 hours is recommended. Persistent negative results can rule out AMI. However, persistent chest pain warrants further testing. Troponins remain elevated for 10-14 days, which may complicate the detection of new infarctions during this period. CK-MB is less sensitive than cTnT or cTnI but is valuable for diagnosing early AMI (<4 hours) and normalizes within 3-4 days. A secondary rise indicates recurrent infarction.
Cardiac biomarkers follow a dynamic pattern, rising to a peak and then returning to baseline. Persistent low-level elevations suggest other causes, such as myocarditis, certain cardiomyopathies, heart failure, and renal dysfunction.
Conventional cardiac enzyme tests, such as creatine kinase (CK), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH), are no longer used for diagnosing AMI due to their lower sensitivity and specificity compared to modern biomarkers.
Diagnosis
The diagnosis of this condition is generally not difficult when based on typical clinical manifestations, characteristic ECG changes, and laboratory findings. For older patients presenting with sudden severe arrhythmias, shock, or heart failure of unknown cause, or with significant and persistent chest tightness, chest pain, or dyspnea, this condition should be considered. It is recommended to initially manage the patient as if they have AMI, and perform dynamic monitoring with ECG and serum cardiac biomarkers over a short period to confirm the diagnosis.
Differential Diagnosis
Angina Pectoris
The fundamental difference lies in the degree of ischemia. Angina is caused by transient myocardial ischemia without necrosis, whereas myocardial infarction involves sustained severe ischemia leading to myocardial necrosis.
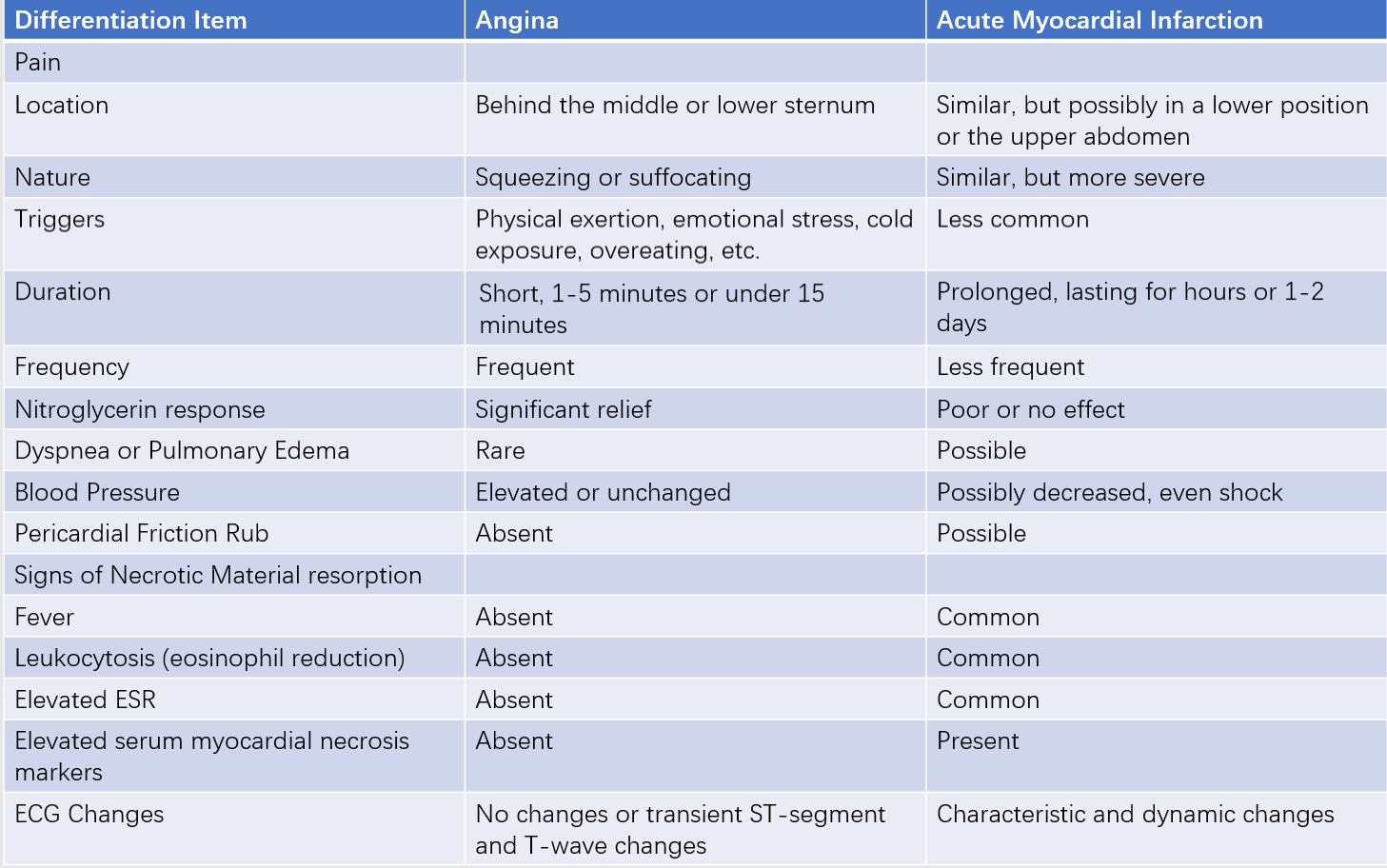
Table 2 Key points for differentiating angina and acute myocardial infarction (AMI)
Aortic Dissection
Chest pain reaches its peak intensity at onset and is described as tearing in nature, often radiating to the back, ribs, abdomen, waist, or lower limbs. Significant differences in blood pressure and pulse may be observed between the two upper limbs. Signs of aortic valve insufficiency may be present, and neurological symptoms such as confusion or hemiplegia may occur. D-dimer levels may be elevated, but serum cardiac biomarkers are typically normal or only mildly elevated. However, Type A aortic dissection involving the coronary ostia may also result in AMI. Echocardiography, chest x-ray, thoracic aortic CTA, or MRA can aid in diagnosis.
Acute Pulmonary Embolism (PE)
Symptoms include chest pain, hemoptysis, dyspnea, and shock, with some patients experiencing syncope. Signs of acute right heart strain may include cyanosis, accentuated P2, jugular vein distention, hepatomegaly, and lower limb edema. ECG changes may include deep S waves in lead I, prominent Q waves and inverted T waves in lead III, leftward shift of the precordial transition zone, and T-wave inversion in right chest leads. Hypoxemia is common, and ventilation-perfusion scans often reveal abnormalities. Echocardiography may show elevated pulmonary artery pressure and acute right heart strain. Pulmonary CTA can detect embolism in large branches of the pulmonary arteries. It is important to note that both AMI and acute PE may present with elevated D-dimer levels, limiting its diagnostic value for differentiation.
Acute Abdominal Conditions
Conditions such as acute pancreatitis, perforated peptic ulcer, acute cholecystitis, and gallstones can cause upper abdominal pain and may be accompanied by shock. A detailed history, physical examination, ECG, and measurements of serum cardiac enzymes and troponins can assist in differentiation.
Acute Pericarditis/Myocarditis
Acute nonspecific pericarditis may present with severe and persistent precordial pain. However, the pain typically coincides with fever, worsens with breathing or coughing, and is accompanied by a pericardial friction rub early on, which disappears along with the pain when pericardial effusion develops. Systemic symptoms are generally less severe than in AMI. ECG changes include ST-segment elevation with a concave upward pattern in most leads except aVR, T-wave inversion, and the absence of abnormal Q waves.
Pheochromocytoma
Sudden elevation of catecholamines may cause headache, palpitations, and perspiration, with significant blood pressure fluctuations. Systolic blood pressure may reach 200 mmHg, followed by a rapid drop, potentially leading to shock. Excessive catecholamine release can cause diffuse coronary vasoconstriction, resulting in myocardial ischemia, ECG ST-segment changes, and elevated cardiac biomarkers, although coronary arteries typically lack significant stenotic lesions. ECG or echocardiographic abnormalities lack localization. Abdominal ultrasound or CT may reveal an adrenal mass, and measurement of catecholamines and their metabolites in blood or urine can aid in diagnosis.
Complications
Dysfunction or Rupture of Papillary Muscle
The overall incidence can reach up to 50%. Ischemia or necrosis of the papillary muscles can impair their contractile function, leading to varying degrees of mitral valve prolapse with regurgitation. This manifests as mid-to-late systolic clicks and a blowing systolic murmur at the apex. The first heart sound may remain unchanged, and heart failure may develop. Mild cases can recover, with the murmur disappearing. Complete rupture of the papillary muscle is rare, usually affecting the posterior papillary muscle in inferior wall MI. This can result in severe heart failure, rapid pulmonary edema, and death within days if surgical intervention is not performed promptly.
Rupture of the Heart
This rare complication typically occurs within the first week after onset and most commonly involves rupture of the free wall of the ventricle, leading to hemopericardium and acute cardiac tamponade, resulting in sudden death. Occasionally, ventricular septal rupture may occur, causing a loud systolic murmur at the left sternal border in the 3rd-4th intercostal space, often accompanied by a thrill. This can lead to heart failure and shock, with death occurring within days. Subacute rupture may allow patients to survive for several months.
Embolism
It occurs in 1%-6% of cases, typically within 1-2 weeks after onset. It is often caused by the detachment of mural thrombi from the left ventricle, leading to arterial embolism in the brain, kidneys, spleen, or limbs. Pulmonary embolism may result from partial detachment of deep vein thrombi in the lower limbs. Large pulmonary emboli can cause sudden death.
Cardiac Aneurysm
Also known as ventricular aneurysm, this condition primarily affects the left ventricle and occurs in 5%-20% of cases. Physical examination may reveal an enlarged left cardiac border and a wide area of cardiac pulsation, sometimes accompanied by a systolic murmur. When mural thrombi form within the aneurysm, heart sounds may be diminished. ECG shows persistent ST-segment elevation. Echocardiography, radionuclide ventriculography, left ventricular angiography, or cardiac MRI may reveal localized protrusion of the ventricular wall with weakened or paradoxical motion. Cardiac aneurysms can lead to heart failure, embolism, and ventricular arrhythmias.
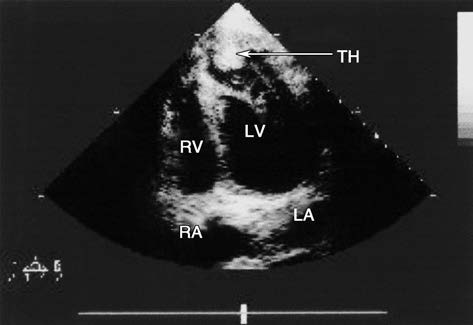
Figure 3 Two-dimensional echocardiography of left ventricular aneurysm (Apical four-chamber view)
The figure shows a left ventricular aneurysm located at the anterior wall and apex, with mural thrombus formation (indicated by the arrow).
LA: Left atrium
LV: Left ventricle
RA: Right atrium
RV: Right ventricle
TH: Thrombus
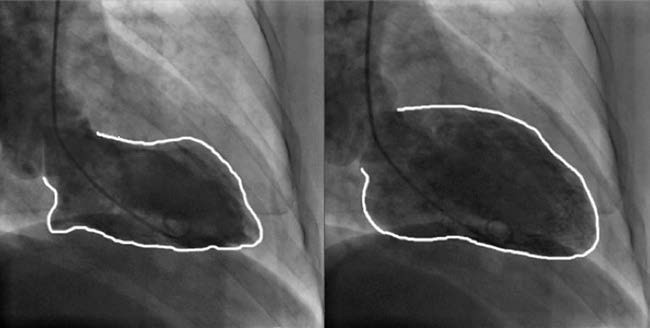
Figure 4 Selective left ventriculography of left ventricular aneurysm
Left panel: Systolic imaging of the left ventricle.
Right panel: Diastolic imaging of the left ventricle.
The apical region shows reduced systolic activity, with a left ventricular ejection fraction (LVEF) measured at 40.1%.
Postmyocardial Infarction Syndrome (Dressler Syndrome)
It occurs in approximately 1%-3% of cases, appearing weeks to months after AMI and potentially recurring. It is characterized by pericarditis, pleuritis, or pneumonitis, with symptoms such as fever and chest pain. The pathogenesis is thought to involve an autoimmune response.
Treatment
Early detection, timely hospitalization, and pre-hospital management are emphasized. The treatment principles focus on rapidly restoring blood flow to the infarcted myocardium (initiating thrombolysis within 30 minutes or percutaneous coronary intervention (PCI) within 90 minutes after first medical contact [FMC]) to salvage ischemic myocardium, prevent infarct expansion, limit the area of myocardial ischemia, preserve and maintain cardiac function, and promptly address severe arrhythmias, pump failure, and various complications to prevent sudden death. The goal is not only to help patients survive the acute phase but also to ensure they retain as much functional myocardium as possible after recovery.
The establishment of a regional emergency network for acute chest pain patients and timely transfer to centers with emergency interventional capabilities significantly impacts patient outcomes.
Monitoring and General Management
During the acute phase, patients should remain on bed rest in a quiet environment. Visits should be minimized to avoid adverse stimuli and alleviate anxiety.
Continuous monitoring of ECG, blood pressure, and respiration should be performed in the coronary care unit (CCU). A defibrillator should always be on standby. For patients with severe pump failure, pulmonary capillary wedge pressure and central venous pressure should also be monitored. Close observation of heart rhythm, heart rate, blood pressure, and cardiac function changes provides objective data for timely therapeutic interventions to prevent sudden death.
For patients with dyspnea and reduced oxygen saturation (<90%), intermittent or continuous oxygen therapy via nasal cannula or mask is recommended during the initial days.
Patients should remain on bed rest for 12 hours during the acute phase. If no complications arise, they should be encouraged to perform limb exercises in bed within 24 hours. By the third day, patients without hypotension can begin walking within the ward. By the fourth or fifth day after infarction, activity can be gradually increased until walking 100-150 meters three times daily. A digestible diet should be provided, and bowel movements should be kept regular.
Maintaining a clear intravenous route for medication administration is essential.
Pain Relief
Myocardial reperfusion therapy, which restores blood supply to ischemic myocardium by opening the infarct-related artery, is the most effective method for pain relief. However, before reperfusion therapy, the following medications can be used to provide rapid pain relief:
- Morphine or pethidine
- Nitrates
- Beta-blockers
Morphine (2-4 mg via intravenous injection) or pethidine (50-100 mg via intramuscular injection) can be administered. If necessary, the dose may be repeated in 5-10 minutes. These drugs can reduce excessive sympathetic nervous system activity and the sense of impending doom. Side effects such as hypotension and respiratory depression should be closely monitored.
Most patients with acute myocardial infarction have indications for nitrate therapy. However, nitrates are contraindicated in patients with inferior wall MI, suspected right ventricular MI, or significant hypotension.
Beta-blockers reduce myocardial oxygen consumption, improve the oxygen supply-demand balance in ischemic regions, limit the size of the infarction, and reduce recurrent myocardial ischemia, reinfarction, ventricular fibrillation, and other malignant arrhythmias. They have been proven to lower mortality during the acute phase. Beta-blockers should be initiated within 24 hours after symptom onset in the absence of the following contraindications:
- Heart failure
- Low cardiac output state
- Increased risk of cardiogenic shock (e.g., age >70 years, systolic blood pressure <120 mmHg, sinus tachycardia >110 bpm, heart rate <60 bpm, or prolonged time since STEMI onset)
- Other contraindications
Cardioselective beta-blockers are generally preferred, starting with a low dose (1/4 of the target dose) and gradually increasing to achieve a resting heart rate of 55-60 bpm.
For patients with severe ischemic chest pain or markedly elevated blood pressure unresponsive to other treatments, intravenous metoprolol may be administered. The recommended dose is 5 mg per injection, repeated every 2-5 minutes as needed. If the heart rate drops below 60 bpm or systolic blood pressure falls below 100 mmHg, the medication should be discontinued. The total intravenous dose should not exceed 15 mg. Oral maintenance therapy should begin 15 minutes after the final intravenous dose.
For patients with relative contraindications to beta-blockers but requiring heart rate reduction, ultra-short-acting intravenous esmolol (50-200 μg/[kg·min]) may be used.
Antiplatelet Therapy
The combined use of oral antiplatelet agents, including aspirin and ADP receptor antagonists, is recommended, starting with a loading dose followed by a maintenance dose. Intravenous glycoprotein IIb/IIIa (GP IIb/IIIa) receptor antagonists are primarily used in patients undergoing direct PCI, administered during the procedure. The choice and usage of antiplatelet agents for STEMI patients are the same as for NSTE-ACS.
Anticoagulant Therapy
Unless contraindicated, all STEMI patients, whether or not they undergo thrombolytic therapy, should routinely receive anticoagulant therapy in combination with antiplatelet therapy. Anticoagulant therapy helps establish and maintain patency of the infarct-related artery and prevents complications such as deep vein thrombosis (DVT), pulmonary embolism (PE), and intracardiac thrombus formation. For direct PCI, particularly in cases with a high risk of hemorrhage, bivalirudin is recommended.
For STEMI patients with intracardiac thrombus or atrial fibrillation, direct oral anticoagulants (DOACs) can be added to antiplatelet therapy, but the risk of hemorrhage should be carefully monitored.
Myocardial Reperfusion Therapy
Reopening the occluded coronary artery within 3-6 hours (or up to 12 hours) after symptom onset to restore myocardial perfusion is one of the most critical treatments for STEMI. This approach salvages myocardium at risk of necrosis, reduces the size of the infarction, and alleviates post-infarction myocardial remodeling.
Recent evidence from clinical trials underscores the importance of timely reperfusion therapy. Establishing regional STEMI management networks and optimizing pre-hospital emergency systems are essential for improving outcomes.
If the patient is in an ambulance or a hospital without percutaneous coronary intervention (PCI) capability but can be transferred to a PCI-capable hospital and undergo PCI within 120 minutes, direct PCI should be the first-choice strategy, aiming to complete reperfusion within 90 minutes.
If the patient is already in a PCI-capable hospital, reperfusion should ideally be completed within 60 minutes.
Indications for primary PCI include:
- Symptoms within 12 hours after onset with persistent ST-segment elevation or new left bundle branch block (LBBB)
- Symptoms between 12-48 hours with evidence of ongoing myocardial ischemia (e.g., persistent chest pain and ECG changes)
Rescue PCI is indicated for patients with persistent chest pain and no significant ST-segment resolution after thrombolytic therapy. Coronary angiography should be performed promptly, and if TIMI grade 0-II flow is observed, indicating failure of artery reperfusion, rescue PCI should be performed immediately.
For patients with successful thrombolysis, emergency coronary angiography and, if necessary, revascularization of the infarct-related artery can relieve ischemia caused by severe residual stenosis and reduce the risk of reinfarction. Stable patients who have undergone successful thrombolysis should ideally undergo coronary angiography within 2-24 hours.
If direct PCI cannot be performed within 120 minutes, thrombolytic therapy should be the first choice, aiming to administer thrombolytic agents within 10 minutes after a definitive ECG diagnosis.
Indications for thrombolysis:
- ST-segment elevation in two or more contiguous leads (≥0.2 mV in chest leads, ≥0.1 mV in limb leads) or a history suggestive of AMI with LBBB within 12 hours after symptom onset, and patient age <75 years
- For patients >75 years old, thrombolysis still considered after careful risk-benefit assessment, with a recommended reduction in thrombolytic drug dosage
- STEMI patients presenting with ongoing ischemic chest pain and widespread ST-segment elevation 12-24 hours after symptom onset
Contraindications for thrombolysis:
- History of hemorrhagic stroke or ischemic stroke within the past 6 months
- Central nervous system damage, intracranial tumor, or vascular malformation
- Recent (2-4 weeks) active internal hemorrhage
- Suspected aortic dissection
- Severe uncontrolled hypertension (>180/110 mmHg) or a history of chronic severe hypertension
- Current use of therapeutic-dose anticoagulants or known hemorrhagic tendencies
- Recent (2-4 weeks) trauma, including head injury, traumatic cardiopulmonary resuscitation, and prolonged CPR (>10 minutes)
- Recent (within 3 weeks) major surgery
- Recent (within 2 weeks) vascular puncture at a non-compressible site
Thrombolytic agents activate plasminogen within the thrombus, converting it into plasmin, which dissolves the fibrin clot in the coronary artery. There are two main types of thrombolytic agents:
- Non-specific plasminogen activators
- Specific plasminogen activators
Non-specific plasminogen activators act on both thrombus-bound and circulating plasminogen, resulting in lower selectivity, reduced reperfusion rates, and higher hemorrhage risks. Examples include urokinase and streptokinase. Streptokinase has antigenicity and cannot be used repeatedly; it is rarely used in modern clinical practice.
Specific plasminogen activators selectively act on plasminogen within the thrombus, minimizing systemic fibrinolytic activity and reducing hemorrhage risks. These are preferred thrombolytic agents.
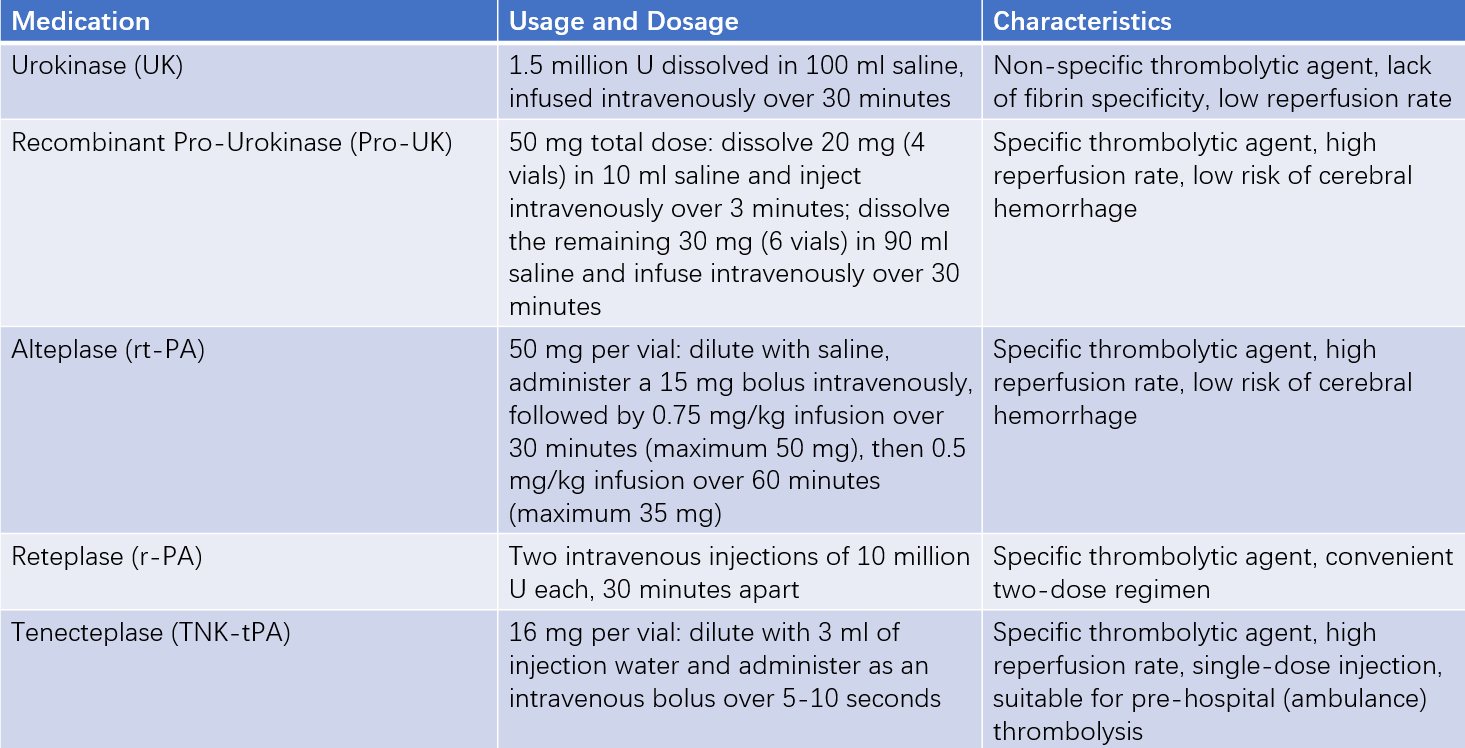
Table 3 Different thrombolytic agents and their usage
Specific thrombolytic agents should be administered alongside heparin-based anticoagulants. For non-specific agents, anticoagulation is required after successful reperfusion. If reperfusion is unsuccessful, rescue PCI should be performed.
Reperfusion success can be directly assessed via coronary angiography (TIMI flow grade II or III) or indirectly through:
- ≥50% resolution of ST-segment elevation within 2 hours
- Significant chest pain relief within 2 hours
- Reperfusion arrhythmias within 2 hours (e.g., transient accelerated idioventricular rhythm, sudden disappearance of AV or bundle branch block, or transient bradycardia or hypotension in inferior wall MI)
- Early peak of serum CK-MB levels (within 14 hours)
For patients with failed PCI or thrombolysis who meet surgical criteria, or those with mechanical complications requiring surgery, emergency coronary artery bypass grafting (CABG) should be performed within 6-8 hours. However, the mortality rate is significantly higher compared to elective CABG.
Acute ischemic myocardial reperfusion may lead to reperfusion injury, often manifesting as reperfusion arrhythmias. Both tachyarrhythmias and bradyarrhythmias may occur, and appropriate rescue measures should be prepared. However, severe arrhythmias are rare, with transient non-paroxysmal ventricular tachycardia being the most common, which typically does not require special treatment.
Renin-Angiotensin-Aldosterone System (RAAS) Antagonists and Angiotensin Receptor-Neprilysin Inhibitors (ARNI)
ACE inhibitors (ACEIs) help improve myocardial remodeling during the recovery phase, reduce mortality, and lower the incidence of congestive heart failure following acute myocardial infarction. They should be used in all patients unless contraindicated.
ARNI, such as sacubitril/valsartan (25-200 mg, twice daily), can be used to inhibit ventricular remodeling after myocardial infarction, reduce the incidence of heart failure, and lower post-MI mortality.
Lipid-lowering Therapy
The use of lipid-lowering drugs, such as statins, is the same as for UA/NSTEMI patients.
Antiarrhythmic and Conduction Disorder Therapy
Arrhythmias must be promptly addressed to prevent progression to severe arrhythmias or sudden cardiac death.
For ventricular fibrillation (VF) or sustained polymorphic ventricular tachycardia (VT), immediate treatment with asynchronous direct current (DC) defibrillation or synchronized DC cardioversion is recommended. Synchronized DC cardioversion should also be used for monomorphic VT if drug therapy is ineffective.
For ventricular premature contractions or VT, lidocaine 50-100 mg can be administered intravenously, repeating every 5-10 minutes until the premature contractions resolve or a total dose of 300 mg is reached, following with a maintenance infusion of 1-4 mg/min. If ventricular arrhythmias recur, amiodarone can be used.
For bradyarrhythmias, atropine 0.5-1 mg can be administered intramuscularly or intravenously.
For second- or third-degree atrioventricular (AV) block with hemodynamic instability, temporary pacing should be performed and discontinued once the conduction block resolves.
For supraventricular tachyarrhythmias, medications such as metoprolol or amiodarone can be used. If drug therapy fails to control the arrhythmia, synchronized DC cardioversion may be considered. Verapamil is generally not recommended during the acute phase of AMI.
Anti-Shock Therapy
Treatment for shock depends on whether it is purely cardiogenic or involves additional factors such as peripheral vascular dysfunction or hypovolemia.
For patients suspected of hypovolemia or with low central venous pressure (CVP) or pulmonary capillary wedge pressure (PCWP), intravenous infusion of dextran 40 or 5-10% glucose solution is recommended. If CVP rises above 18 cmH2O or PCWP exceeds 15-18 mmHg after infusion, further fluid administration should be stopped. Elevated CVP in right ventricular infarction is not a contraindication for volume resuscitation.
If blood pressure remains low after volume resuscitation but PCWP and cardiac index (CI) are normal (indicating inadequate peripheral vascular tone), vasopressors such as dopamine [starting dose 3-5 μg/(kg·min)] or norepinephrine (2-8 μg/min) can be administered. Dobutamine [starting dose 3-10 μg/(kg·min)] is another option for intravenous infusion.
If blood pressure remains low despite the above measures, with elevated PCWP, low CI, or significant peripheral vasoconstriction (manifesting as cold extremities and cyanosis), intravenous sodium nitroprusside can be started at 15 μg/min, gradually increasing the dose every 5 minutes until PCWP decreases to 15-18 mmHg. Alternatively, intravenous nitroglycerin can be started at 10-20 μg/min, with dose increments every 5-10 minutes until left ventricular filling pressure decreases.
Additional measures include correcting acidosis, avoiding cerebral ischemia, protecting renal function, and using digitalis preparations if necessary. To reduce the mortality of cardiogenic shock, hospitals with the necessary resources can consider using intra-aortic balloon counterpulsation (IABP) or left ventricular assist devices (LVADs) for circulatory support.
Anti-Heart Failure Therapy
The primary focus is on treating acute left heart failure, primarily using morphine (or pethidine) and diuretics. Vasodilators can also be selected to reduce left ventricular load, or positive inotropic agents may be used. Digitalis preparations should be used with caution as they may induce ventricular arrhythmias. In patients with right ventricular infarction within the first 24 hours after the onset of myocardial infarction, diuretics should be used cautiously.
Management of Right Ventricular Myocardial Infarction
The treatment approach differs slightly from that of left ventricular infarction. In cases where right ventricular infarction causes right heart failure with hypotension but without signs of left heart failure, volume expansion is recommended. Intravenous fluid infusion should be administered under hemodynamic monitoring until hypotension is corrected or pulmonary capillary wedge pressure reaches 15 mmHg. If hypotension persists after 1-2 liters of fluid infusion, positive inotropic agents, preferably dobutamine, should be used. Diuretics are not recommended. Temporary pacing may be performed in cases of accompanying atrioventricular (AV) block.
Other Therapies
The following treatments may help salvage jeopardized myocardium, prevent infarct expansion, reduce the ischemic area, and accelerate healing. However, these therapies are not yet fully established or remain controversial, and their use should be considered based on the specific condition of the patient.
In the early phase, if there are signs of sympathetic overactivity such as tachycardia and beta-blockers are contraindicated, calcium channel blockers such as diltiazem may be considered. These can lower heart rate, reduce myocardial oxygen consumption, and limit the size of the infarct. However, attention should be paid to potential side effects such as hypotension and negative inotropic effects. Routine use of calcium channel blockers in AMI patients is not recommended.
For patients who cannot tolerate beta-blockers or have a persistently elevated sinus heart rate despite the maximum tolerated dose of beta-blockers, ivabradine may be used to lower the sinus heart rate and reduce myocardial oxygen consumption.
A solution containing 1.5 g of potassium chloride, 10 U of insulin, and 500 ml of 10% glucose can be administered via intravenous infusion 1-2 times daily for 7-14 days as a treatment course. This therapy promotes myocardial uptake and metabolism of glucose, facilitates the intracellular movement of potassium ions, restores the polarized state of the cell membrane, supports normal cardiac contraction, and reduces arrhythmias.
Rehabilitation and Post-Discharge Treatment
Rehabilitation therapy is recommended after recovery from AMI. After 2-4 months of physical activity training, patients may gradually resume partial work or reduce their workload. Some patients may eventually return to full-time work, but they should avoid heavy physical labor or excessive mental stress.
Prognosis
The prognosis depends on factors such as the size of the infarct, the development of collateral circulation, and the timeliness of treatment. Approximately half of all AMI-related deaths occur before hospital admission, with a significant proportion of out-of-hospital deaths attributed to sudden cardiac death.
Due to advancements in coronary care units, modern pharmacological therapies, and reperfusion strategies, the in-hospital mortality rate for AMI patients has decreased from 30% in the past to approximately 4% in centers performing direct PCI. Most deaths occur within the first week, especially within the first few hours, with a particularly high mortality rate in patients who develop severe arrhythmias, shock, or heart failure.
Prevention
Primary prevention involves preventing atherosclerosis and coronary artery disease in the general population. Secondary prevention focuses on preventing recurrent infarction and other cardiovascular events in patients with a history of coronary artery disease or myocardial infarction.