Hypertrophic cardiomyopathy (HCM) is a type of cardiomyopathy characterized by hypertrophy of the left and/or right ventricle along with diastolic dysfunction. It is necessary to exclude myocardial hypertrophy secondary to increased afterload or systemic diseases. The incidence of HCM is approximately 2‰ in adults and 0.02-0.05‰ in children, with a prevalence of 0.29‰. Clinical manifestations vary greatly, and in some patients, sudden cardiac death (SCD) may be the first presentation, making HCM a significant cause of sudden death in adolescents and athletes.
Pathogenesis
HCM is an autosomal dominant genetic disorder, with about half of cases being familial. Mutations in at least eight sarcomeric protein-encoding genes have been identified. Approximately 60% of patients have pathogenic or likely pathogenic variants, with mutations in MYH7 and MYBPC3 accounting for 70% of cases. In the remaining 40% of patients without detectable genetic variants, the condition is often sporadic. The diverse phenotypes of HCM are influenced by interactions between genetic and environmental factors.
Pathology
Anatomical Features
Most cases exhibit left ventricular hypertrophy, particularly asymmetric hypertrophy of the interventricular septum. In some patients, the hypertrophy occurs in atypical regions.
Pathological Features
Disorganized myocardial cell alignment, fibrosis, scar formation, and small vessel disease are characteristic findings.
Pathophysiology
The primary pathophysiological mechanisms of HCM include left ventricular outflow tract obstruction (LVOTO) and diastolic dysfunction:
Thickening of the interventricular septum, with or without mitral valve abnormalities (e.g., elongated leaflets, anterior displacement of the valve and papillary muscles), can lead to structural LVOTO. During systole, high-velocity blood flow through the obstructed area creates a suction effect, causing systolic anterior motion (SAM) of the mitral valve leaflets, which can further exacerbate functional obstruction.
Mitral regurgitation may occur secondary to SAM or due to primary valvular disease.
Myocardial cell hypertrophy, fibrosis, and abnormal calcium reuptake during diastole can reduce ventricular compliance and lead to diastolic dysfunction.
Myocardial thickening, reduced microvascular density, and impaired coronary flow reserve can result in myocardial ischemia.
HCM patients may exhibit abnormal heart rate and blood pressure responses during exercise, such as a failure of systolic blood pressure to increase by >20 mmHg or a paradoxical drop in systolic blood pressure >20 mmHg at peak exercise.
Clinical Manifestations
Symptoms
Dyspnea is primarily exertional, but may also occur at rest. About 1/3 of patients experience exertional chest pain or tightness. Arrhythmias are common in most patients, but symptomatic arrhythmias mainly include atrial fibrillation (AF), ventricular tachycardia (VT) [including sustained and non-sustained VT (SVT/NSVT)], and even ventricular fibrillation (VF). Heart failure (HF) is typically seen in the middle to late stages of the disease, presenting with symptoms of heart failure with preserved ejection fraction (HFpEF) or reduced ejection fraction (HFrEF). Syncope/sudden cardiac death is often triggered by exercise. About 1/6 of patients experience at least one episode of syncope, and SCD may be the first symptom in some cases.
Signs
Mild cardiac enlargement can be seen. A fourth heart sound may be heard. A systolic ejection murmur is audible at the left sternal border in the 3rd-4th intercostal spaces. A systolic blowing murmur may be heard at the apex.
The intensity of the murmur is variable. Maneuvers or medications that increase myocardial contractility or decrease preload (e.g., positive inotropic agents, standing, or sublingual nitroglycerin) can enhance the murmur, whereas the murmur diminishes with maneuvers or medications that reduce myocardial contractility or increase preload.
Auxiliary Examinations
Electrocardiography (ECG)
The main ECG findings include high QRS voltage, ST-segment depression, T-wave inversion, abnormal Q waves, intraventricular conduction block, and arrhythmias.
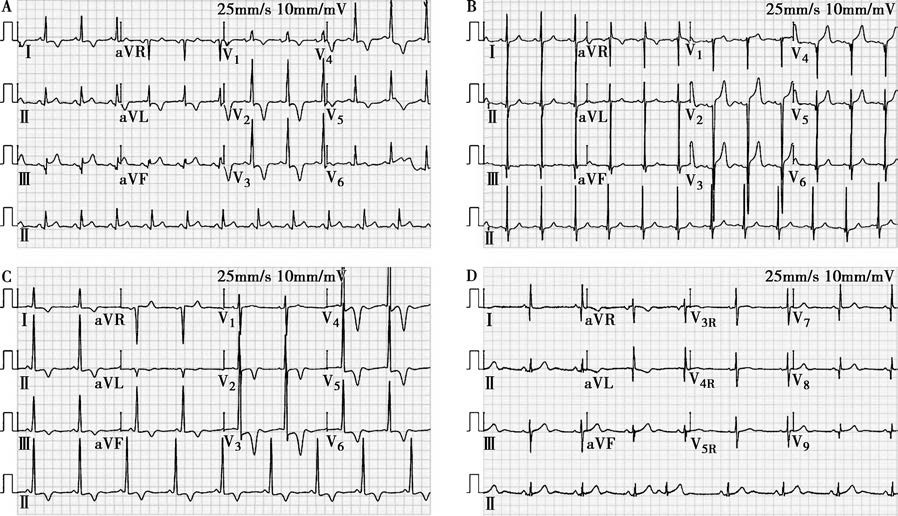
Figure 1 Electrocardiographic manifestations of hypertrophic cardiomyopathy
A. A 56-year-old male patient with obstructive hypertrophic cardiomyopathy involving the left ventricular outflow tract. Abnormal q waves are observed in leads II, III, and aVF, along with T-wave inversion in leads I, aVL, and V1-V5.
B. A 23-year-old female patient with mid-septal obstructive hypertrophic cardiomyopathy. Abnormal q waves are seen in leads II, III, aVF, and V3-V6, with poor R-wave progression in leads V1-V2.
C. A 74-year-old female patient with apical hypertrophic cardiomyopathy. Widespread T-wave inversion is observed, particularly fixed and symmetric T-wave inversion in the left precordial leads.
D. An 81-year-old female patient with non-obstructive hypertrophic cardiomyopathy. Narrow and deep abnormal q waves are noted in leads I, aVL, and V4-V9 (leads V4-V6 are not shown).
Characteristic features are:
- Fixed ST-segment depression and T-wave inversion, distinguishing it from the dynamic changes seen in myocardial ischemia
- Fixed deep and symmetric T-wave inversion, indicative of apical hypertrophy
- Pathological Q waves that are deep but not wide, appearing in leads inconsistent with coronary artery distribution, distinguishing them from infarction-related Q waves
Echocardiography
Transthoracic echocardiography (TTE) is the first-choice diagnostic tool and is used for initial evaluation, regular follow-up, family screening, and assessing treatment efficacy. The main findings on TTE include asymmetric hypertrophy of the interventricular septum, left ventricular outflow tract obstruction, systolic anterior motion (SAM) of the mitral valve, and left ventricular diastolic dysfunction.
Key points to note:
- Hypertrophy can occur in any region of the myocardium
- If no obstruction is detected at rest, a provocation test should be performed. This involves placing the patient in a semi-supine position and using the Valsalva maneuver during Doppler imaging. If LVOTO with a pressure gradient ≥50 mmHg is not induced, an exercise stress test (rather than pharmacological stress) should be conducted.
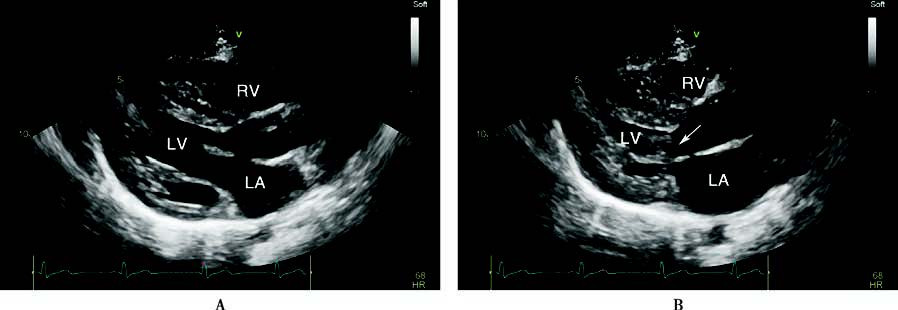
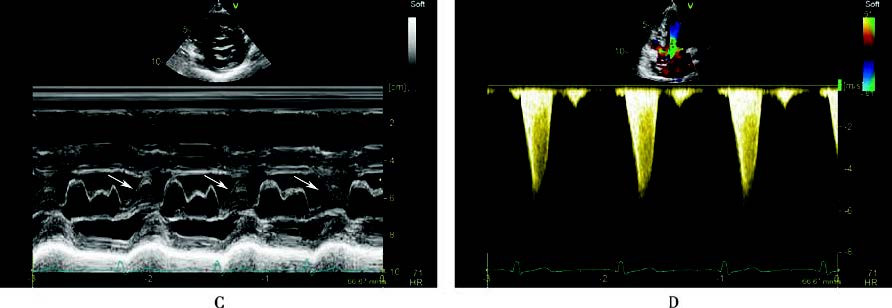
Figure 2 Echocardiographic findings in hypertrophic cardiomyopathy
A 56-year-old male patient with left ventricular outflow tract hypertrophic obstructive cardiomyopathy. The long-axis and short-axis views of the left ventricular outflow tract showed significant thickening of the left ventricular wall, with marked thickening of the interventricular septum (A, B, C). In the end-systolic phase, two-dimensional echocardiography (B) and M-mode echocardiography (C) demonstrated anterior motion of the anterior leaflet of the mitral valve, which, together with the hypertrophied interventricular septum, caused outflow tract obstruction (indicated by arrows). Continuous-wave Doppler echocardiography in the apical five-chamber view measured a peak pressure gradient of 144 mmHg in the left ventricular outflow tract (D).
LA: Left atrium
LV: Left ventricle
RV: Right ventricle
Cardiac Magnetic Resonance (CMR)
CMR is highly accurate for assessing the thickness, location (especially the apex or right ventricle), and degree of fibrosis in hypertrophied myocardium. Extensive late gadolinium enhancement (LGE) (≥15% of left ventricular mass) indicates a high risk of sudden cardiac death. However, CMR is time-consuming and expensive, making it more suitable for cases where TTE findings are inconclusive.
Endomyocardial Biopsy (EMB)
Endomyocardial biopsy can help differentiate HCM from other types of cardiomyopathies.
Genetic Testing
Family screening and genetic testing are valuable for differential diagnosis, prognosis assessment, and reproductive decision-making. First-line testing should target the eight sarcomeric genes (MYH7, MYBPC3, TNNI3, TNNT2, TPM1, MYL2, MYL3, ACTC1). If results are negative, exome sequencing may be considered. Genetic screening of first-degree relatives is recommended at any age. For genotype-positive but phenotype-negative individuals, repeat screening every 2-3 years is advised until the age of 50.
Diagnosis and Differential Diagnosis
Imaging evidence of myocardial wall thickening is essential for diagnosing HCM. It is necessary to exclude myocardial hypertrophy caused by increased cardiac afterload or systemic/metabolic diseases. EMB and genetic testing may be performed when needed.
Diagnostic criteria in adults include:
- In the absence of other causes, a diastolic wall thickness of ≥15 mm in any part of the left ventricle on imaging
- For individuals with a family history or positive genetic test, a wall thickness of ≥13 mm
Diagnostic criteria in children include:
- In asymptomatic children without a family history, a left ventricular wall thickness z-score (number of standard deviations from the mean) >2.5
- For children with a family history or positive genotype, a z-score >2
Diagnostic criteria in relatives include:
- For adult first-degree relatives, a left ventricular wall thickness >13 mm
- For pediatric first-degree relatives, a z-score <2 with morphological or ECG abnormalities suggestive of a diagnosis but not sufficient for a definitive diagnosis
Classification
Classification based on hemodynamics:
- Obstructive type: Peak pressure gradient at the obstruction site ≥30 mmHg, including both resting and dynamic obstruction
- Non-obstructive type: Peak pressure gradient at the obstruction site <30 mmHg at rest or during provocation
Classification based on location of hypertrophy:
- Septal hypertrophy: The most common type, primarily involving the basal interventricular septum
- Apical hypertrophy: Primarily affecting the apex and near-apical regions
- Mid-ventricular hypertrophy: Involving the ventricular wall corresponding to the papillary muscles
- Right ventricular hypertrophy: Rare
Differential Diagnosis
HCM must be distinguished from more common causes of left ventricular hypertrophy due to increased afterload, such as hypertension, aortic valve stenosis, and athlete’s heart. It is also necessary to rule out systemic diseases that can cause myocardial hypertrophy, such as amyloidosis, sarcoidosis, Gaucher disease, glycogen storage disorders, Fabry disease, Danon disease, and hemochromatosis.
Treatment
Current treatment options for obstructive hypertrophic cardiomyopathy have not been definitively proven to alter the natural course of the disease. Therefore, the primary goals of treatment are to alleviate symptoms, reduce complications, and prevent sudden cardiac death.
Pharmacological treatment for symptomatic obstructive HCM
Beta-Blockers
Beta-blockers, such as metoprolol and bisoprolol, are the first-line treatment. They exert negative chronotropic and inotropic effects, which help reduce LVOTO, lower ventricular filling pressures, and improve symptoms. Treatment should start at a low dose and be titrated gradually to the effective or maximum tolerated dose (target resting heart rate: 55-60 bpm).
Myosin Inhibitors
Mavacamten is currently the only approved myosin inhibitor. It selectively inhibits the ATPase activity of myosin heavy chains, reversibly suppresses excessive cross-bridge formation between actin and myosin, reduces myocardial hypercontractility, and improves myocardial compliance and energy metabolism. It is indicated for symptomatic adult patients with NYHA class II-III obstructive HCM. However, it is contraindicated in patients with a left ventricular ejection fraction (LVEF) <55%.
Calcium Channel Blockers
Non-dihydropyridine calcium channel blockers, such as verapamil and diltiazem, have negative chronotropic and inotropic effects. They reduce obstruction, improve ventricular filling, and alleviate symptoms. These agents can be used as alternatives for patients who cannot tolerate beta-blockers or for whom beta-blockers are contraindicated.
Other Medications
Class Ia antiarrhythmic drugs, such as disopyramide and procainamide, may be used in patients with severe symptoms of obstructive HCM that persist despite adequate treatment with beta-blockers and non-dihydropyridine calcium channel blockers.
To avoid exacerbating LVOTO, the following should be avoided: dehydration, excessive alcohol consumption, arterial vasodilators (e.g., dihydropyridine calcium channel blockers), high doses of diuretics, positive inotropic agents, and intravenous vasodilators.
Pharmacological treatment for HCM with comorbidities or complications
Concurrent Heart Failure
Treatment strategies for HCM patients with heart failure (including HFpEF and HFrEF) should follow general heart failure management guidelines.
Concurrent Atrial Fibrillation
In patients with HCM and AF, anticoagulation therapy is recommended regardless of the CHA2DS2-VASc score, provided there are no contraindications.
Surgical treatment for symptomatic obstructive HCM
Septal reduction therapy (SRT) includes percutaneous or intramyocardial septal ablation and surgical septal myectomy.
Indications for SRT are:
- Clinical criteria: Severe symptoms (NYHA class III-IV) despite optimal medical therapy, or severe activity-related symptoms (e.g., recurrent syncope) related to LVOTO
- Obstruction criteria: Resting or provoked LVOTO peak pressure gradient ≥50 mmHg associated with septal hypertrophy or SAM
SRT is not recommended for asymptomatic obstructive HCM patients or those with symptoms adequately controlled by optimized medical therapy.
Myocardial Ablation
This includes alcohol septal ablation (ASA) and transthoracic intramyocardial radiofrequency ablation. Complications include conduction block, myocardial infarction, and scar-mediated ventricular arrhythmias, with an incidence <2%.
Surgical Septal Myectomy
Compared to myocardial ablation, surgical myectomy provides more significant relief of LVOTO and clinical symptoms. Complications include conduction block, atrial fibrillation, ventricular septal perforation, and aortic regurgitation, with a procedure-related mortality rate <1%.
Cardiac Pacing
Artificially altering ventricular activation sequences can reduce obstruction and symptoms, but the efficacy remains uncertain.
Heart Transplantation
This is the last resort for patients with end-stage HCM.
Exercise Recommendations
Genotype-positive but phenotype-negative individuals may participate in competitive sports. Most HCM patients can engage in low- to moderate-intensity recreational activities. Patients with severe obstructive HCM should avoid competitive sports.
SCD Prevention
An implantable cardioverter-defibrillator (ICD) is an absolute indication for survivors of SCD (secondary prevention) and a relative indication for high-risk patients (primary prevention). The decision to implant an ICD depends on the patient’s preference, predicted survival (≥1 year), and risk stratification. These indications are generally applicable to all forms of cardiomyopathy.
For HCM patients, guidelines recommend using the HCM Risk-SCD model for risk stratification.
Clinical risk factors for SCD include:
- Early-onset family history of SCD
- History of cardiac arrest or hemodynamically unstable sustained ventricular tachycardia
- Exercise-induced or unexplained syncope
- Severe left ventricular hypertrophy (≥30 mm)
- Apical aneurysm
- LVEF <50%
- Recurrent non-sustained VT (NSVT)
- Extensive late gadolinium enhancement (LGE) ≥15% of left ventricular mass