Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a myocardial disease characterized by progressive atrophy of the right ventricular myocardium, which is replaced by fibrous and/or fatty tissue. The prevalence of this disease is approximately 0.2‰ - 1.0‰, with a male predominance. It typically manifests around the age of 30 and has a poor prognosis.
Pathogenesis
ARVC is mostly a familial autosomal dominant disorder, often accompanied by variable expressivity and incomplete penetrance. To date, multiple genetic loci have been identified, with at least 10 well-established pathogenic genes. The majority are genes encoding desmosomal proteins, including plakophilin-2 (PKP2), desmoplakin (DSP), desmoglein-2 (DSG2), desmocollin-2 (DSC2), and junction plakoglobin (JUP). There are also non-desmosomal protein-coding genes associated with ARVC.
The exact pathogenesis remains unclear, but ARVC is currently considered a desmosomal disease. Non-desmosomal genes may contribute to the disease by affecting desmosomal function. Additionally, inflammatory responses are thought to play a role in disease progression.
Clinical Features
Arrhythmias
Arrhythmias are the most common clinical manifestation, presenting as progressive palpitations, tachypnea, and syncope. They are often triggered by emotional stress or physical exertion. The hallmark arrhythmias include premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), with left bundle branch block (LBBB) morphology sustained or non-sustained VT (SVT/NSVT) being particularly characteristic.
Sudden Cardiac Death (SCD)
In some patients, SCD is the first manifestation of ARVC, particularly in young individuals aged ≤35 years. These patients are often asymptomatic before the event, with emotional stress and intense physical activity being the main triggers.
Heart Failure (HF)
HF typically presents in the late stages of the disease, usually in patients over 40 years old, with predominant symptoms of right-sided heart failure.
Diagnostic Workup
Electrocardiogram (ECG)
Repolarization and depolarization abnormalities, as well as arrhythmias, are key diagnostic indicators. Routine surface ECG or signal-averaged ECG may show T-wave inversion in the right precordial leads and Epsilon waves. Ventricular arrhythmias, particularly LBBB morphology SVT/NSVT, have both diagnostic and prognostic significance.
Transthoracic Echocardiography (TTE) / Cardiac Magnetic Resonance (CMR)
TTE and CMR are the primary tools for assessing cardiac structure and function. Characteristic findings include reduced or paradoxical motion of the right ventricular wall, bulging of the right ventricular wall, and reduced right ventricular ejection fraction (RVEF). CMR with late gadolinium enhancement (CMR-LGE) can detect fibrous and/or fatty replacement in the myocardium of any ventricle.
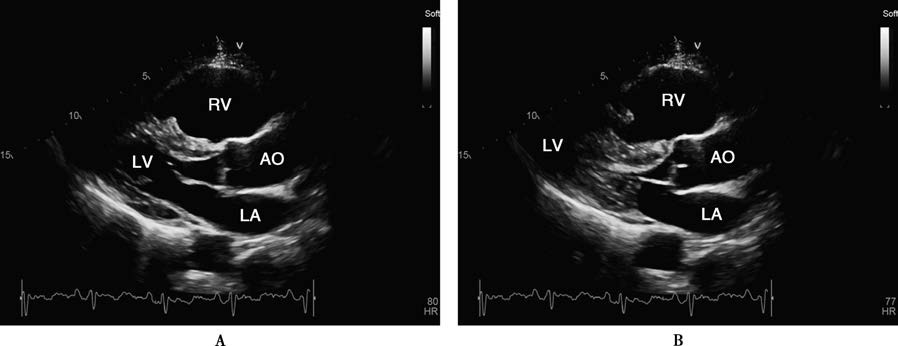
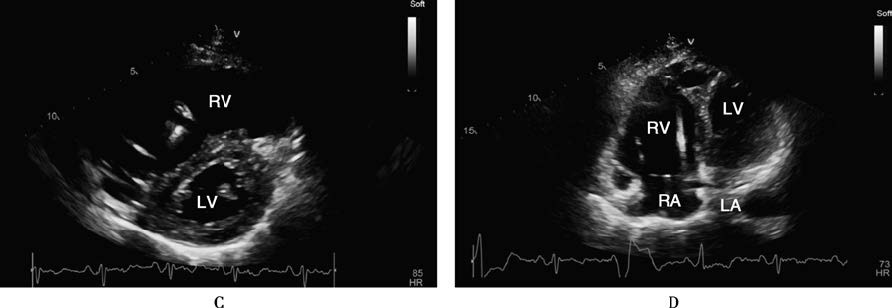
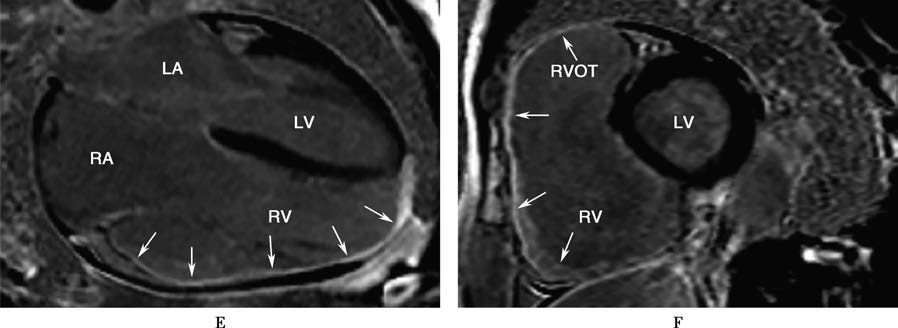
Figure 1 Transthoracic echocardiography and cardiac magnetic resonance imaging findings in arrhythmogenic right ventricular cardiomyopathy
A 70-year-old male patient with ARVC who had an ICD implanted. Transthoracic echocardiography (TTE) in the parasternal long-axis view of the left ventricle during diastole (A) and systole (B), short-axis view (C), and apical four-chamber view (D) shows severe right ventricular dilation, thinning of the ventricular wall, and globally reduced wall motion. Cardiac magnetic resonance imaging (CMR) with gadolinium contrast using a phase-sensitive inversion recovery (PSIR) sequence in the four-chamber view (E) and short-axis view of the left ventricle (F) reveals thinning of the right ventricular wall and extensive fibrous tissue replacement (indicated by arrows).
Abbreviations: AO, Aorta; LA, Left Atrium; LV, Left Ventricle; RA, Right Atrium; RV, Right Ventricle; RVOT, Right Ventricular Outflow Tract.
Genetic Testing
Pathogenic or likely pathogenic mutations can be identified in up to 60% of patients. However, the penetrance of these mutations depends on factors such as age, sex, and physical activity. Therefore, the pathogenicity of genetic variants should be repeatedly evaluated.
Diagnosis
ARVC should be suspected in patients with right ventricular enlargement and LBBB morphology VT. Diagnosis is based on a combination of clinical, electrocardiographic, and imaging findings. Endomyocardial biopsy (EMB) and genetic testing may be required in certain cases.
Major diagnostic criteria include:
- Structural/functional abnormalities: TTE or CMR showing diffuse or localized right ventricular wall motion abnormalities or aneurysms, with RVEF ≤ 40%
- Tissue abnormalities: CMR showing fibrous and/or fatty replacement of the right ventricular free wall myocardium
- Repolarization abnormalities: T-wave inversion in the right precordial leads
- Depolarization/conduction abnormalities: Epsilon waves in the right precordial leads
- Arrhythmias: LBBB morphology SVT/NSVT
- Family history: A first-degree relative meeting diagnostic criteria, confirmed by autopsy or surgical pathology, or carrying a pathogenic or likely pathogenic gene.
Differential Diagnosis
Uhl anomaly, also known as parchment right ventricle, is a rare congenital condition characterized by the absence of right ventricular myocardium, leaving only the endocardium and epicardium. It primarily affects infants and is not familial, with early death from HF being common.
Idiopathic right ventricular outflow tract tachycardia is a benign ventricular tachycardia originating from the right ventricular outflow tract, with no family history and a good prognosis.
Treatment
The primary goals of treatment are to manage HF and arrhythmias, alleviate symptoms, and reduce the risk of SCD.
Antiarrhythmic drugs, such as beta-blockers, sotalol, amiodarone, and verapamil, can be used empirically. A combination of amiodarone and beta-blockers may also be effective.
For patients with severe right ventricular dysfunction and refractory ventricular arrhythmias, heart transplantation may be considered.
Sudden Cardiac Death Prevention
ICD implantation is the mainstay therapy for SCD survivors or high-risk patients. However, the decision to implant an ICD should take into account the patient’s preferences and predicted survival (≥1 year).
High-risk factors include:
- Survivors of SCD
- Sustained or non-sustained VT with syncope or hemodynamic instability
- QRS dispersion ≥ 40 ms
- Severe right ventricular dilation, RVEF < 40%, or left ventricular involvement with LVEF < 45%, as confirmed by imaging
- Severe early symptoms, including presyncope or syncope
- Carriers of high-risk pathogenic genes for SCD